Правило аддитивности как прогнозирование численного значения энергии высшей заполненной молекулярной орбитали
Математически строго аддитивны только массы смешиваемых тел, но иногда аддитивные объемы, а также молекулярные массы.
Касаемо структуры молекул, правило аддитивности может быть использовано для установления строения молекул. Расхождения до 0,2 - 0,4 см3 относятся за счет возможных ошибок опыта и неточности самих аддитивных констант. У ионных соединений небольшие отклонения от аддитивности связаны с взаимной поляризацией ионов. Полезно также при определении состава комплексов.
Правило аддитивности ковалентных связей справедливо только для несопряженных связей. При сопряжении длины связей сильно отклоняются от стандартных значений; двойные связи удлиняются, а простые связи между ними укорачиваются.
Правило аддитивности атомных радиусов не выполняется только при сильном различии химической природы двух атомов в соединении.
Для постановки эксперимента были использованы органические производные фосфина. При этом для первого этапа исследования, который включал в себя определение значение энергии HOMO основных функциональных групп, применялись органические молекулы (ОМ), являющиеся монозамещенными фосфинами РН2-СН2-FG и РН2-FG, где FG -- функциональная группа. Проведение второго этапа исследования включало в себя В качестве проверки правила аддитивности были использованы структуры состава:
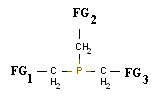
.
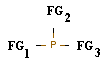
Функциональные группы выбирались по их способности к проявлению +М (доноргые) или - М (акцепторные) характера по принципу последовательного чередования их в молекуле.
Квантово-химический расчет (численный эксперимент) был проведен в программе GAMESS-2009, силами средств визуализации входной структуры программного комплекса CambridgeSoft 2013 и выходных данных по граничным орбиталям программы Chemissian 4.33. ОМ были предварительно оптимизированы сначала методом ММ+, далее методом Ab Initio HF/MINI*. Собственно расчет производился методом Ab Initio МР2/DZV*.
В качестве упрощения, было принято, что Е (HOMO) - РН2 = Е (HOMO)РН3, а также, что Е (HOMO) - СН3 = const = 0.634, что было вычислено на основе метилфосфина.
Энергия HOMO молекулы фосфина РН3 равна -10,482 eV. Ниже приведены энергии HOMO основных функциональных групп (табл. 1):
Табл. 1. Энергии HOMO основных функциональных групп
|
Функциональная группа |
Вклад в HOMOОМ, eV |
Функциональная группа |
Вклад в HOMOОМ, eV |
|
-O- |
0,452 |
-OH |
0,433 |
|
-СI |
-0,051 |
-COOH |
0,044 |
|
Вторичный атом углерода* |
0,068 |
-CN |
-0,354 |
|
-NH- |
-0.095 |
-S- |
0.615 |
|
-С ОС- |
-0,019 |
-CH=CH2 |
0,710 |
|
-SO3H |
-0,378 |
-C6H5 |
1,585 |
|
-NH2 |
0,531 |
-NH-N=O |
-0,021 |
|
-СН=О |
-0,032 |
-СО- |
0,234 |
|
-N-/ |
-0.481 |
-P-/ |
-0.389 |
*имеется в виду - СН2-; радикал метилен =СН2 дает вклад в HOMOОМ +0,604 eV.
Очевидно, что структуры, имеющие отрицательный знак, дают уменьшение энергии HOMOОМ.
Проведение второго этапа вычислений по проверке правила аддитивности проводилось согласно выведенной автором эмпирической формуле:
Где -- Е (HOMO) "неорганического прототипа" (НП: в рамках статьи РН3)
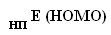
-- Е (HOMO) функциональной группы в органической молекуле
-- Е (HOMO) "центрального фрагмента" (ЦФ: в рамках статьи третичный атом фосфора), порождаемого НП
Результаты представлены в таблице 3 для ОМ состава
Согласно справочным данным, взятым из таблицы 1
Табл. 2. Сведения о проверке правила аддитивности для несопряженных молекул
|
№ п/п ОМ |
№ п/п FG |
Наименование FG |
Значение энергии HOMOОМ, eV |
Разница "т--п", eV |
K, теор/прак | |
|
Практическое* |
Теоретическое | |||||
|
1 |
FG1 FG2 FG3 |
|
-9,434 |
-9,553 |
0,119 |
1,013 |
|
2 |
FG1 FG2 FG3 |
|
-10,210 |
-10,291 |
0,081 |
1,008 |
|
3 |
FG1 FG2 FG3 |
|
-9,448 |
--9,382 |
0,066 |
0,993 |
|
4 |
FG1 FG2 FG3 |
|
-9,845 |
-10,040 |
0,195 |
1,020 |
* Имеется в виду проведенное в программе GAMESS-2009. Теоретическое -- проведенное по правилу аддитивности.
Видно, что разница между теоретически и практически вычисленными значениями энергии превышает 0,1 только в случае с 2 донорными и 1 акцепторным заместителем. Наиболее надежен вывод формулы -- с коэффициентом пересчета, основанным на отношении (k). Видно, что в ОМ, где заместители являются сходными по свойствам (все донорные или все акцепторные), коэффициент возможно принять как 1,01. В ОМ, где заместители являются различными по свойствам коэффициент возможно принять как 1.006, хотя в этом случае установленное значение коэффициента менее надежно.
Результаты представлены в таблице 3 для ОМ состава
,
Где реализуется явление сопряжения или существенного уменьшения прочности связи Р-- FG, согласно справочным данным, взятым из таблицы 1
Табл. 3. Сведения о проверке правила аддитивности для молекул, проявляющих явление сопряжения или существенного уменьшения прочности связи Р-- FG
|
№ п/п ОМ |
№ п/п FG |
Наименование FG |
Значение энергии HOMOОМ, eV |
Разница "т--п", eV |
K, теор/прак | |
|
Практическое |
Теоретическое | |||||
|
1 |
FG1 FG2 FG3 |
|
-10,006 |
-9,553 |
0,453 |
0,955 |
|
2 |
FG1 FG2 FG3 |
|
-11,173 |
-10,291 |
0,882 |
0,921 |
|
3 |
FG1 FG2 FG3 |
|
-9,565 |
--9,382 |
0,183 |
0,981 |
|
4 |
FG1 FG2 FG3 |
|
-10,370 |
-10,040 |
0,330 |
0,968 |
Очевидно, что разница между теоретически и практически вычисленными значениями энергии превышает 0,4 только в случае с ОМ, имеющей заместители акцепторного характера, причем числовое значение очень близко к 1 eV. Очевидно, что в ОМ №№ 1, 3, 4 коэффициент k возможно принять в среднем как 0,97. В ОМ № 2, коэффициент принимает усредненное значение 0,9 .
У несопряженных соединений в целом Е (HOMO) меньше.
Таким образом, связь между двумя методами расчета энергии высшей заполненной молекулярной орбитали (практическим: с помощью квантовохимического пакета PCGAMESS2009 и теоретическим: по правилу аддитивности) имеет вид:
Химический атомный аддитивность
,
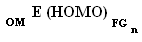
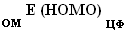
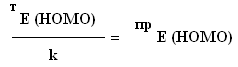
Где k - численное значение коэффициента
Правильность выведения автором справочных данных, несмотря на некоторые допущения, подтверждается крайне малым отличием теоретически вычисленных значений от практически. Правило аддитивности как инструмент прогнозирования численного значения энергии высшей заполненной молекулярной орбитали, может быть использовано в случае несопряженных и сопряженных соединений с точностью до 0,1. В этом случае данные таблицы 1 могут претендовать на универсальность в случае, когда необходимо провести расчет значения энергии высшей заполненной молекулярной орбитали для органических молекул, произошедших от неорганического низкомолекулярного соединения (простых эфиров, аминов и др.). Весьма вероятно, что вышесказанное относится и к значению энергии низшей вакантной молекулярной орбитали, а также для молекул, не имеющих родственного генетически связанного с ними неорганического низкомолекулярного соединения.
Похожие статьи
-
Зависимость скорости реакции от температуры определяется правилом Вант-Гоффа : При повышении температуры на каждые 10о скорость большинства реакций...
-
1. В результате линейной комбинации две атомные орбитали (АО) формируют две молекулярные орбитали (МО) - связывающую, энергия которой ниже, чем энергия...
-
Эконометрическое моделирование и прогнозирование объемов таможенных платежей в регионе деятельности Ростовской таможни В настоящее время для...
-
Эконометрика (задания выполнить в ППП Excel, по каждому пункту сделать выводы) Рассмотреть экономическое явление, в котором участвуют 2 фактора...
-
Расположение пунктов питания и потребленияэлектрической энергии - Районная электрическая сеть
Таблица 1 - Сведения о потребителях электроэнергии по пунктам Наименование данных Пункты 1 2 3 4 5 6 Наибольшая зимняя активная нагрузка, МВт 12 14 16 26...
-
ПОНЯТИЕ ОБ АВТОКОРРЕЛЯЦИИ. ОПРЕДЕЛЕНИЕ СИЛЫ АВТОКОРРЕЛЯЦИИ Парные регрессионные модели отражают специфику взаимодействия некоторого функционального...
-
Способность минералов закрепляться на поверхности раздела воздух - вода (или в общем случае газ - жидкость) зависит от степени полярности минеральной...
-
Построение корреляционных моделей исследуемых явлений
Построение корреляционных моделей исследуемых явлений Цель работы: На основе данных статистических наблюдений вывести корреляционные зависимости в виде...
-
Рис. 5. Масс-спектрометрия электронной ионизацией (EI). Электронная ионизация - один из наиболее важных способов ионизации для повседневных анализов...
-
Результаты исследований А. Н. Фрумкина и Б. В. Дерягина показали, что характер изменения удельной поверхностной энергии е прослоя воды, находящегося...
-
Собственно-корреляционные параметрические методы изучения связи - Основы эконометрики
Измерение тесноты и направления связи является важной задачей изучения и количественного измерения взаимосвязи социально-экономических явлений. Оценка...
-
Распределением признака Называется закономерность встречаемости разных его значений. Нормальное распределение Характеризуется тем, что крайние значения...
-
Методы прогнозирования в статистике населения - Система источников данных о населении
Моделирование временного тренда среднегодовой численности занят Ого населения Санкт-Петербурга Приведем данные среднегодовой численности занятого...
-
В первоначальном выборе объясняющих переменных существует две стратегии. Часть авторов осуществляют подбор переменных, опираясь на собственные...
-
На следующем этапе в модель были добавлены дамми-переменные годов и отраслей. Таблицы соотношения переменных и данных приведены ниже. Кроме дамми...
-
Реализуем математическую модель (2) (6) в MS Excel. Для этой цели построим таблицы исходных данных задачи по расчету оптимального графика занятости при...
-
Связь phenoxy гербицидов с саркомами мягких тканей и лимфомами типа нон-Ходкина - Токсичные вещества
Саркомы мягких тканей и лимфомы нон-Ходкина связывают с воздействием phenoxy гербицидов с 1970-х гг. после ряда докладов в Швеции. При изучении...
-
Опис-ся правиломВант-Гоффа: С увелич-ем темп. на каждые 10 градусов. Скор. больш-ва хим. р-ций увелич-ся в 2-4 раза. Где г-темп. коэф-т скорости хим....
-
Построение многофакторной корреляционно-регрессионной модели производительности труда
Построение многофакторной корреляционно-регрессионной модели производительности труда Данная работа направлена на выявление факторов, от которых зависит...
-
Класс алкилбензолов представлен 37 соединениями в табл.3, где приведены экспериментальные данные, использованные нами при определении значений параметров...
-
Регрессия -- зависимость среднего значения какой-либо величины от некоторой другой величины или от нескольких величин. Задача регрессионного анализа...
-
Экономический корреляционный регрессионный Парная линейная регрессия Парная регрессия характеризует связь между двумя признаками: результативным и...
-
Описание используемых методов - Моделирование вероятности банкротства
В данной работе было принято решение использовать логистический анализ с помощью пакета STATA, а также алгоритм CART с помощью SPSS Modeler. Бинарная...
-
В интервальном вариационном ряду мода вычисляется по формуле: , Где Y O - нижняя граница модального интервала; H - размер модального интервала; F Mo -...
-
Основные понятия и определения проблемы прогнозирования - Прогнозирующие системы
Необходимо отметить, что мы рассматриваем прогнозирование в целях планирования производства или управления запасами. Таким образом, наш интерес лежит в...
-
Прогнозирование курса Ukb/Usd, Общий подход к прогнозированию курса UKB/USD - Прогнозирующие системы
В данной главе описаны эксперименты по прогнозированию курса американского доллара по отношению к украинскому карбованцу (UKB/USD). Сначала описаны...
-
В данной главе описан способ прогнозирования с помощью НС, основанный на методе окон. Также приведен обзор применения НС в финансовой сфере. Общий подход...
-
Строение, физические свойства, значение воды - Свойства водорода
Вода -- самое распространенное соединение водорода. Общая масса воды на нашей планете около 1,4*1018 т. Вода -- это единственное вещество, все три...
-
Аннотация - Точность математического прогнозирования как функция количества учитываемых факторов
В статье рассмотрена точность прогнозирования экономических показателей в зависимости от количества используемых параметров на основе математического...
-
Ответ: а) выбрать число, имеющее наименьшее количество десятичных знаков. B) Другое число округлить на один десятичный знак больше выбранного. С)...
-
Из истории комбинаторики - Правила комбинаторики
Комбинаторика занимается различного вида соединениями, которые можно образовать из элементов конечного множества. Некоторые элементы комбинаторики были...
-
Химия 20 века - Этапы становления химии
Конец 19 в. ознаменовался тремя выдающимися открытиями в области физики, в результате которых была доказана сложная структура атома, прежде считавшегося...
-
Среди различных конфигураций искусственных нейронных сетей встречаются такие, при классификации которых по принципу обучения, строго говоря, не подходят...
-
Методы изучения связи качественных признаков - Основы эконометрики
При наличии соотношения между вариацией качественных признаков говорят об их ассоциации, взаимосвязанности. Для оценки связи в этом случае используют ряд...
-
Применение статистических методов анализа для адекватной интерпретации результатов контроля остаточных знаний соискателей высшего образования на примере...
-
При использовании статистических методов прогнозирования во многих случаях необходимо знать возможную ошибку прогноза, т. е. тот интервал, в котором...
-
Теперь, когда в рамках данного исследования была получена модель с наилучшими характеристиками для непубличных строительных компаний, полученные...
-
МЕТОДЫ ОТБОРА СПЕЦИАЛИСТОВ В ЭКСПЕРТНУЮ ГРУППУ - Основы прогнозирования
Проведение экспертизы начинается с создания специальной группы специалистов-организаторов опроса. Задачами группы являются выбор цели экспертизы,...
-
Достоинства, сложности и недостатки модели Леонтьева - Многосекторные модели прогнозирования
"Модель В. В. Леонтьева имеет ряд очевидных достоинств: - наличие простых вычислительных алгоритмов; - возможность информационного обеспечения (на основе...
-
Введение, Методы экстраполяции - Формализованные методы прогнозирования
К формализованным методам относятся методы экстраполяции и методы моделирования. Они базируются на математической теории. Среди методов экстраполяции...
Правило аддитивности как прогнозирование численного значения энергии высшей заполненной молекулярной орбитали