Металлорганосилоксаны в катализе - Современные тенденции развития металлоорганосилоксанов
Традиционно органосиланоляты щелочных металлов использовали как катализаторы анионной полимеризации органоциклосилоксанов для получения линейных полимерных органосилоксанов. В дальнейшем было обнаружено, что применение смешанных силанолят-алкоголятов K4Li4(OSiMe3)6-8(OBuT)1-2 Позволяет синтезировать высокомолекулярные органосилоксаны с низкой полидисперсностью.
В большинстве случаев каталитические свойства МОС обусловлены природой металла, представляющего собой каталитический центр. Силоксигруппы, как правило, играют второстепенную роль, позволяя соответствующим образом сформировать каталитический центр; однако в некоторых случаях участие силоксановой матрицы становится определяющим.
Ряд работ посвящен исследованию каталитических свойств МОС в реакции конверсии алкенов. Так, были созданы катализаторы, имитирующие катализаторы Циглера-Натты. В этих системах сочетается комплекс переходного металла с AlR3. Например, показано, что спироциклические V(IV)- и Cr(II)-органосилоксаны (соединения 44 и 45 соответственно) совместно с AlMe3 Катализируют полимеризацию этилена и пропилена.

Более оригинальна идея использовать органосилоксановый каркас в роли силикатной матрицы, применяемой обычно в качестве неорганического носителя каталитически активных центров. Например, полиэдрический Cr(VI)-органосилоксан 46 по каталитической активности не уступает классическому катализатору Филлипса (Cr/SiO2).

Современные исследования МОС-катализаторов для конверсии алкенов ориентированы в первую очередь на использование низкокоординационных комплексов. Некоторые триорганосилоксипроизводные металлов (особенно трехкоординационное соединение вольфрама 7) представляют интерес как перспективные катализаторы, активирующие алкены и алкины:


Диорганосилоксигидридотитан (активированный триметилфосфином) образует интермедиат с этиленом и катализирует превращение, приводящее к образованию бут-1-ена и высших олигомеров этилена.

В некоторых случаях генерирование алкена происходит при конверсии алкилсилоксипроизводного металла: так, при термолизе соединения вольфрама 47 получаются алкены в результате в-элиминирования алкильных групп, одновременно с этим образуются алкеновые комплексы.

Наиболее отчетливо преимущество объемных триорганосилоксизаместителей у атомов металла проявилось при изучении каталитических свойств Nb - и Ta-органосилоксанов. Следует отметить, что именно с этих металлов Ричард Шрок (лауреат Нобелевской премии по химии 2005 г.) начинал поиски катализаторов реакции метатезиса, вводя в окружение металлического центра Трет-бутоксигруппы, чтобы придать стабильность получаемым из них металлокарбенам. Более поздние исследования показали, что той же цели можно достичь иным путем - с помощью Трет-бутилсилоксигрупп, причем с эффективным результатом, поскольку к каталитическому центру удается присоединить большее число объемных органических групп. Установлено, что триорганосилоксипроизводные ниобия и тантала способны присоединять алкены с образованием металлокарбенов, катализирующих реакции метатезиса.


К другому типу катализаторов относятся триорганосилоксизамещенные гидриды тантала, способные присоединять СО, что позволяет рассматривать их как перспективные катализаторы реакции Фишера-Тропша.

Полиэдрические TiIII- и TiIV-силсесквиоксаны, представляющие собой замкнутые (соединение 48) или частично замкнутые (соединение 1) полиэдры, используют как гомогенные или гетерогенные катализаторы эпоксидирования алкенов.

Триорганосилоксипроизводные платиновых металлов находят традиционное применение, определяемое природой металлического центра. Роль органосилоксигрупп - повышение стабильности катализаторов при многократном использовании. Триорганосилоксипроизводное родия [Ph3SiORh(CO)2]2 - эффективный катализатор гидроформилирования.

В процессах гидрирования СО комплексы RMOSiPh3?2 L (M = Pt, Pd; R = Me, Et, Ph; L - циклоокта-1,5-диен) более эффективны по сравнению с соединениями R2M ? 2L или H2PtCl6/SiO2.
При каталитическом действии комплексов Pd в процессах кросс-сочетания с участием кремнийорганических реагентов постулируются 163 Образование фрагмента Si-O-Pd в интермедиате.
Олигомерные МОС также применяют в катализе. Следует отметить, что использование их в качестве связующих для наполненных материалов малоперспективно, поскольку в большинстве случаев они образуют полимерные системы со сравнительно невысокой молекулярной массой (2000-5000), не обладающие конструктивной прочностью. В то же время изучение их каталитических свойств привело к интересным и в некоторых случаях неожиданным результатам.
Достаточно очевидная идея использовать эти соединения в качестве аналогов неорганических силикатов обусловила выбор катализируемых реакций, прежде всего процессов нефтехимического синтеза. Было установлено, что при высокотемпературной поликонденсации олиго-МОС постепенно теряют органические группы, превращаясь в неорганические силикаты, но при этом сохраняют особенности структуры исходных МОС. Такие соединения успешно конкурируют с близкими по составу катализаторами - неорганическими силикатами, - однако МОС обладают более высокой активностью и селективностью в процессах крекинга, галогенирования, дегидратации, алкилирования и др.
В дальнейшем олиго-МОС стали применять в таких каталитических реакциях, где металлосиликаты практически не используют. Примером может служить стереоселективное окисление углеводородов (в частности, 1,4-диметилциклогексана) в присутствии олигоферрофенилсилоксана.

Наиболее важное применение олиго-МОС нашли в конверсии галогенуглеводородов. Эти процессы лежат в основе целого ряда многотоннажных производств (получение поливинилхлорида, дихлорэтана, хлорбензола, хлоралканов и т. д.). Своеобразие галогенуглеводородов заключается в низкой склонности к координации с большинством металлокомплексов, что затрудняет подбор катализаторов. Выбор был основан на следующем соображении: наиболее эффективный катализ следует ожидать в тех случаях, когда становится возможным радикально-цепной механизм с участием ионов металлов переменной валентности. Переходные металлы, входящие в структуру органосилоксановой матрицы, могут изменять степень окисления (ключевой момент катализа для указанного класса реакций), оставаясь в составе силоксанового скелета. Были приняты во внимание и некоторые дополнительные достоинства олиго-МОС: их растворимость в органических растворителях позволяла использовать из в гомогенном катализе, а в случае гетерогенного катализа их можно химически связать с широко применяемыми минеральными носителями (SiO2, Al2O3 и др.). Кроме того, доступны системы с участием почти любых переходных металлов или их сочетаний в заданном соотношении. Варьирование их свойств может быть реализовано также путем изменения параметров высокотемпературных конденсационных процессов, влияющих на формирование поверхности образующегося катализатора.
Были исследованы 166 каталитические свойства олигомерных Cu-, Co - и Fe - органосилоксанов в изомеризации несимметричного 3,4-дихлорбут-1-ена в симметричный 1,4-дихлорбут-2-ен (ключевая стадия в процессе производства хлоропропенового каучука). Наибольшую каталитическую активность показал Cu-содержащий МОС, что подтвердило существующие представления о частичном восстановлении ионов металла в ходе каталитической реакции - среди исследованных металлов наиболее легко восстанавливается ион меди. Тем не менее Co - и Fe-содержащие олиго-МОС также представляют интерес, поскольку их действие более селективно: в их присутствии не происходит образование Цис-изомера 1,4-дихлорбут-2-ена. Главным отличием олиго-МОС от традиционных катализаторов (нафтената или галогенида меди) оказалась исключительно высокая стабильность: многократное их использование (6-10 раз) не приводит к заметному снижению активности, в то время как традиционные катализаторы практически полностью теряют активность после первого применения.
Были изучены каталитические свойства олиго-МОС в процессах обменного галогенирования, позволяющих получать галогенуглеводороды без использования галогенов в виде простых веществ.

В случае хлоруглеводородов данный метод позволяет осуществить экологически более предпочтительное производство. Это особенно актуально при получении бромпроизводных, поскольку прямое бромирование молекулярным бромом протекает с трудом и неселективно; кроме того, получение бромидов обычно представляет собой многостадийный процесс.
Детальное изучение 148,167,168 обменного галогенирования углеводородов n-C10H22, n-C12H26, cyclo-C6H12, P-Me2C6H4 И C6H5Me с участием CCl4 или CBr4 (в присутствии каталитических количеств олиго-Cu-фенилсилоксана) позволило выяснить механизм процесса. Реакция протекает по радикальному механизму: передача по цепочке радикального центра от радикала ?ССl3 К углеводороду показана в верхней ветви схемы, превращение каталитического центра показано в нижней ветви (согласно классическим представлениям, катализатор после участия в реакции возвращается в исходное состояние, что отмечено изогнутой стрелкой). Активность олиго-Cu-фенилсилоксана практически не снижается после пятикратного использования, в то время как стандартный катализатор CuCl2 ? (DMF)N Теряет работоспособность в течение одного цикла.
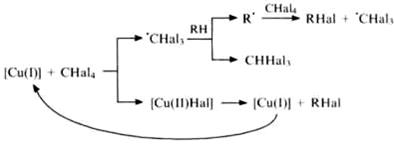
Рассмотренная реакция обменного галогенирования приводит к смеси моногалогенпроизводных с различным расположением атома галогена. Процесс присоединения галогенуглеводородов к алкенам более селективен, поскольку в этом случае положение двойной связи указывает место присоединения атома Cl и фрагмента CCl3.

В качестве катализаторов этой реакции были исследованы монофенил - и дифенилсодержащие олигомерные Cu-органосилоксаны. Процесс протекает по радикальному механизму, подобному описанному выше. Отличие состоит в том, что образование радикального центра при участии двойной связи алкена облегчено. Показано, что у структурированных (содержащих каркасные фрагменты) монофенилпроизводных, включающих трифункциональные фрагменты RSiO1.5, доступность каталитических центров и, соответственно, каталитическая активность ниже, чем у дифенилпроизводных, построенных их линейных фрагментов.
Кинетическое изучение двух указанных типов процессов галогенирования позволило установить, что заметная часть каталитических центров оказывается "выключенной" из каталитического процесса из-за того, что значительная доля ионов металла связана в межцепные координационные кластеры. Металл стремится заполнить свою координационную сферу, привлекая атомы кислорода соседних фрагментов Si-O-M.

В результате ион металла оказывается координационно насыщенным и структурно малодоступным для взаимодействия с CCl4. Таким образом, основная задача - затруднить образование межцепных металлооксидных кластеров. Если блокировать металл каким-либо лигандом, то это затормозит межцепную координацию, но не поможет решить основную проблему - лиганд также станет затруднять приближение реагента. Было найдено решение, достаточно простое для реализации, - заменить связанную с кремнием фенильную группу нонильной. При этом ион Cu в составе олигононилсилоксана оказывается (по данным УФ-спектроскопии) координационно ненасыщенным.
Таким образом, объемная алифатическая группа эффективно затрудняет координационное взаимодействие атомов меди в соседних цепях, "укрывая" металлические центры, но в реакционной среде она обладает подвижностью полимерного сегмента и не препятствует приближению органического реагента (CCl4) к каталитическому центру (рис.1).

Результаты реализации этой идеи достаточно убедительны: при каталитическом присоединении CCl4 К окт-1-ену активность нонилсодержащих полимерных Cu-МОС вдвое выше, чем у медьфенилсилоксанов.
Рассмотренный в обзоре материал убедительно показывает, что металлоорганосилоксаны представляют собой класс соединений, чрезвычайно интересный и с теоретической, и с практической точек зрения.
Похожие статьи
-
Интерес к ПМОС в немалой степени был вызван рядом практически полезных свойств, которыми они обладают: высокая термическая и термоокислительная...
-
Одним из наиболее актуальных направлений полимерной химии в настоящее время является создание новых композиционных материалов. С этой точки зрения...
-
МОС несомненно интересны в плане создания полимерных нанокомпозитов, представляющих собой полимерную матрицу с диспергированными в ней нанометровыми...
-
Для исследования процесса деструктивного сульфидирования МОС при их взаимодействии с сероводородом выбраются соединения с сендвичевым типом структуры:...
-
В 1947 году Андрианов К. А. показал, что кремнийорганические соединения, содержащие функциональные гидроксильные группы у атома кремния (диэтилсиландиол,...
-
Синтез гибридных органо-неорганических нанокомпозиционных материалов -- новое ответвление в золь-гель науке и технологии. Первые работы по синтезу...
-
Металлоорганосилоксаны (МОС) - достаточно распространенное семейство соединений, характеризующихся наличием структурной группировки...
-
ПМОС - аморфные вещества, и поэтому однозначное определение их структуры является очень сложной задачей. Тем не менее, на основании анализа их...
-
Для проведения исследований физико-химических характеристик образцов применялся ряд физико-химических методов. Молекулярно-массовое распределение...
-
Структура каркасных металлоорганосилоксанов - Современные тенденции развития металлоорганосилоксанов
За последние десятилетия был синтезирован и охарактеризован сравнительно большой ряд разнообразных каркасных МОС. Каркасные МОС имеют удивительную...
-
НЕМНОГО О ПРОМЫШЛЕННОМ КАТАЛИЗЕ - Процесс катализа
На всю жизнь запомнилась мне проводившаяся по Энглеру разгонка полученного конденсата, в котором уже в начале опыта бензиновая фракция составляла 67%. Мы...
-
Определение тангенса (tg д) диэлектрических потерь и диэлектрической проницаемости (е) проводится на Q-метре фирмы "Тесла", при частоте 1000 Гц. Для...
-
В работах авторов [16, 17] исследованы кремнийорганические полимерные композиции, структурированные сульфидами переходных металлов (Cu, Zn), обладающий...
-
Большое значение координационные соединения приобрели в качестве катализаторов промышленно важных процессов. Запатентовано огромное число различных...
-
Введение, Гомогенный и гетерогенный катализ - Гомогенный и гетерогенный катализ
Химические реакции протекают с различными скоростями. Некоторые из них полностью заканчиваются за малые доли секунды, другие осуществляются за минуты,...
-
Химическая технология и тенденции ее развития - Этапы становления химии
Потребности общества породили химическую технологию. По выражению Бертло, химия начинает творить свой собственный объект исследования, создавая сотни...
-
Катализ - Концепции современного естествознания: химическая составляющая
Наиболее сильное влияние на скорость реакции оказывает присутствие в реагирующей системе Катализатора -- вещества, которое повышает (а иногда и уменьшает...
-
Гетерогенный катализ - каталитические реакции, идущие на поверхности раздела фаз, образуемых катализатором и реагирующими веществами. Механизм...
-
ГЕТЕРОГЕННЫЙ КАТАЛИЗ - Процесс катализа
К сожалению, до сих пор, несмотря на достаточно большое число теорий и гипотез в области катализа, многие основополагающие открытия были сделаны случайно...
-
Гидрирование Большое число каталитических реакций связано с активацией атома водорода и какой-либо другой молекулы, приводящей к их химическому...
-
Носитель катализатора, иначе подложка (катализатора) (англ. carrier или support) -- инертный или малоактивный материал, служащий для стабилизации на его...
-
Высокая каталитическая эффективность. Отличительной особенностью любого фермента является его чрезвычайно высокая каталитическая эффективность. Так,...
-
Энергетический профиль каталитической реакции - Гомогенный и гетерогенный катализ
Энергетический профиль реакции - это кривая, которая показывает зависимость координаты реакции (насколько прошла реакция) от времени (при постоянном...
-
Гетерогенный катализ - Гомогенный и гетерогенный катализ
Механизм гетерогенного катализа включает пять стадий, причем все они обратимы. 1. Диффузия реагирующих веществ к поверхности твердого вещества. 2....
-
Гомогенный катализ - Гомогенный и гетерогенный катализ
Среди многочисленных каталитических реакций особое место занимает катализ в цепных реакциях. "Цепными реакциями, как известно, называются такие...
-
ГОМОГЕННЫЙ КАТАЛИЗ - Процесс катализа
Среди многочисленных каталитических реакций особое место занимает катализ в цепных реакциях. "Цепными реакциями, как известно, называются такие...
-
До недавнего времени об эволюционной химии ничего не было известно. В отличие от биологов, химиков не интересовал вопрос о "происхождении видов"...
-
Современная картина химических знаний - Естественнонаучные концепции развития химических знаний
Современную картину химических знаний объясняют с позиций четырех концептуальных систем, которые схематично представлены на рис.1. Рис.1. На рисунке...
-
РОЛЬ КАТАЛИЗА В ЭКОЛОГИИ - Процесс катализа
Огромную роль призван сыграть катализ в решении актуальнейшей проблемы - охраны окружающей среды. По словам Кусто, земной шар напоминает "одиноко...
-
Активность и селективность катализатора - Гомогенный и гетерогенный катализ
Все процессы переработки углеводородного сырья сопровождаются фазообразованием (испарение, катализ, крекинг и др.). Одна из задач физико-химической...
-
"Катализ. Гомогенный и гетерогенный катализ" - Гомогенный и гетерогенный катализ
В 1835 году Берцелиус впервые использовал термин "катализатор" для обозначения "веществ, которые способны пробудить сродство, дремлющее при данной...
-
1. Copyright © С. И. Левченков, 1996 - 2005. 2. Шмидт Ф. К. Физико-химические основы катализа -- И.: Фрактал -- 2004. -- С. 9 3. Ходаков Ю. В., Эпштейн...
-
Гомогенный и гетерогенный катализ. Механизм действия катализатора - Основы химии
При гомогенном катализе катализатор и реагирующие вещества образуют одну фазу (газовая смесь или раствор). При гетерогенном катализе катализатор и...
-
ВИДНЫЕ ДЕЯТЕЛИ ХИМИИ О КАТАЛИЗЕ - Процесс катализа
И. Берцелиус (1837): "Известные вещества оказывают при соприкосновении с другими веществами такое влияние на последние, что возникает химическое...
-
Катамлиз (греч. кбфЬлхуйт восходит к кбфблэейн -- разрушение) -- избирательное ускорение одного из возможных термодинамически разрешенных направлений...
-
КАТАЛИЗ В БИОХИМИИ - Процесс катализа
Ферментативный катализ неразрывно связан с жизнедеятельностью организмов растительного и животного мира. Многие жизненно важные химические реакции,...
-
КАТАЛИЗ - процесс, заключающийся в изменении скорости химических реакций в присутствии веществ, называемых катализаторами. Катализаторы - вещества,...
-
Превращение аренов - Каталитический риформинг
Каталитический риформинг алкан газ Незамещенные соединения в условиях процесса риформинга устойчивы. Метилзамещенные арены (толуол, ксилолы) подвергаются...
-
Выявление основной тенденции развития В ходе обработки динамического ряда важнейшей задачей является выявление основной тенденции развития явления...
-
Система химии, логика ее развития и построения Что такое химия? Химия является высокоупорядоченной - постоянно развивающейся системой знаний о веществах,...
Металлорганосилоксаны в катализе - Современные тенденции развития металлоорганосилоксанов