Третье начало термодинамики - Физическая химия
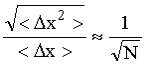
Третий закон термодинамики (постулат Планка). Термодинамические потенциалы. Энергия Гельмгольца и энергия Гиббса как критерии направленности и предела протекания процессов. Зависимость энергии Гельмгольца и энергии Гиббса от параметров состояния. Уравнения Гиббса-Гельмгольца. Расчет изменения стандартных энергий Гиббса и Гельмгольца в химических реакциях при различных температурах.
Система с переменным составом. Химический потенциал. Общие условия равновесия в системах с переменным составом.
Ранее внутренняя энергия рассматривалась в виде суммы двух величин "свободной" и "связанной" энергии. Возможность рассчитать величину "свободной" энергии, т. е. той части внутренней энергии системы, которую можно превратить в работу, дает тепловая теория Нернста, называемая также третьим началом термодинамики.
Положения теории Нернста заключаются в следующем:
- 1. При абсолютном нуле температуры свободная энергия Есв равна теплоте процесса. 2. При температурах, близких к абсолютному нулю, теплоемкость системы равна нулю.
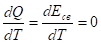
Одной из формулировок третьего начала термодинамики является также постулат Планка:
Энтропия идеального кристалла при абсолютном нуле температуры равна нулю.
Есть и другое определение 3-его закона термодинамики:
Существует экстенсивная функция состояния термодинамической системы - энтропия (S). При протекании в изолированной системе обратимых процессов эта
Функция остается неизменной, а при необратимых - увеличивается
Строго говоря, тепловая теорема Нернста и постулат Планка являются следствиями из второго начала термодинамики.
Расчет абсолютной энтропии
Условия расчета: изменение энтропии происходит при нагревании системы от абсолютного нуля до температуры T при постоянном давлении. Из первого и второго начал термодинамики имеем:
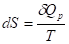
Отсюда
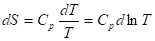
Учитывая, что ST=0 = 0, получим:
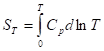
При T = 0 любое вещество может находиться только в твердом состоянии. При нагревании вещества возможен его переход в жидкое и затем в газообразное состояние; для фазовых переходов, происходящих в изобарно-изотермических условиях, изменение энтропии равно приведенной теплоте фазового перехода:
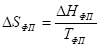
Если рассматривать нагревание вещества без фазовых переходов, то оно сопровождается непрерывным ростом энтропии, в то время как при фазовом переходе происходит скачкообразное изменение энтропии.
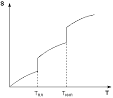
Рис Зависимость энтропии вещества от температуры.
Учитывая это, рассчитать абсолютную энтропию любого вещества при любой температуре можно следующим образом:
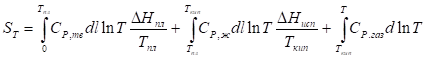
Так как изменение энтропии в ходе химического процесса определяется только видом и состоянием исходных веществ и продуктов реакции и не зависит от пути реакции; то:
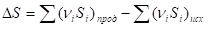
Расчет изменения энтропии в различных процессах
Расчеты изменения энтропии в различных процессах основаны на использовании соотношения дQ = TdS и частных производных энтропии по термодинамическим переменным:
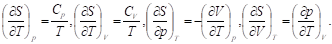
Последние два тождества представляют собой соотношения Максвелла.
Изменение энтропии при изменении температуры
Процесс Изменение энтропии
Изохорное нагревание/охлаждение
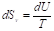
; ;
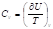
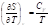
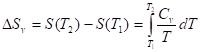
Изобарное
Нагревание/охлаждение
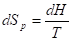
; ;
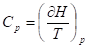
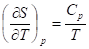
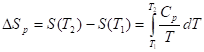
Изотермическое расширение/сжатие
Выражение через изменение объема
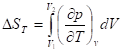
;
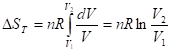
Выражение через изменение давления
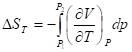
;
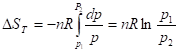
Фазовый переход
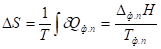
Т. к. при плавлении (кипении) происходит поглощение теплоты, то энтропия в этих процессах возрастает: SТв < SЖ < SГ..
Если температура фазового перехода не равна температуре обратимого фазового перехода, то использовать это уравнение нельзя, так как при необратимых процессах:
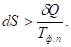
Изотермически-изобарное смешение идеальных газов
При смешивании n1 молей одного газа объемом V1, с другим в количестве n2 молей и объемом V2 общий объем будет (V1 + V2), причем газы расширяются независимо друг от друга и занимают весь объем, поэтому общее изменение энтропии равно сумме изменений энтропии каждого газа:
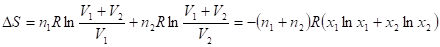
Где xi - мольная доля i-го газа в полученной газовой смеси. Изменение энтропии в данном случае всегда положительно, т. к. все ln xi < 0, поэтому идеальные газы всегда смешиваются необратимо. Если при аналогичных условиях смешивать две порции одного и того же газа, то это уравнение уже неприменимо. Никаких изменений в системе при смешивании не наблюдается, и ДS = 0. Тем не менее, в формуле для расчета ДS не содержится никаких индивидуальных параметров газов, поэтому, казалось бы, должна быть применима и к смешению одинаковых газов. Это противоречие называют парадоксом Гиббса.
Химическая реакция Разность мольных энтропий продуктов реакции и реагентов, взятых в стандартных состояниях, называется стандартной энтропией реакции. Для реакции
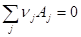
Стандартная энтропия реакции равна разности абсолютных энтропий продуктов и реагентов с учетом стехиометрических коэффициентов:
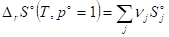
Для расчета абсолютной энтропии веществ в стандартном состоянии надо знать зависимости теплоемкости Cp от температуры для каждой из фаз, а также температуры и энтальпии фазовых переходов.
Пример:
Абсолютная энтропия газообразного вещества в стандартном состоянии при температуре T складывается из следующих составляющих:
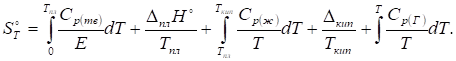
Абсолютные энтропии участников реакции при давлении, отличном от стандартного, находят следующим образом:
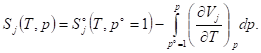
Характеристические функции. Термодинамические потенциалы.
Внутренняя энергия и энтропия относятся к классу характеристических функций. Функция называется характеристической, если все термодинамические свойства гомогенной системы могут быть выражены непосредственно через нее и ее частные производные по соответствующим переменным. Эти независимые переменные называют естественными. Характеристические функции, по определению, содержат в себе всю термодинамическую информацию о системе. Но некоторые из естественных переменных, например, энтропию, нельзя измерять (контролировать) в ходе какого-то процесса. Поэтому встает задача перехода от одних переменных к другим - экспериментально измеримым, но с условием сохранения характеристичности самой функции. Такой переход осуществляют с помощью преобразований Лежандра.
С помощью этих преобразований вводятся другие характеристические функции (в скобках указаны их естественные переменные):
- * энтальпия H(S, p, n) = U + pV, * энергия Гельмгольца F(T, V, n) = U - TS, * энергия Гиббса G(T, p, n) = U + pV - TS = H - TS = F + pV.
Функции U, H, F, G называют также термодинамическими потенциалами. Зависимость термодинамических потенциалов от их естественных переменных описывается основными уравнениями термодинамики - фундаментальными уравнениями Гиббса:
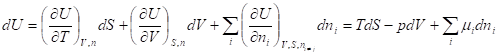
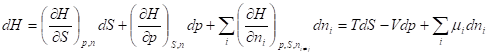
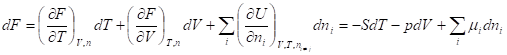
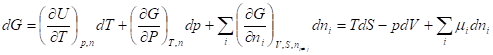
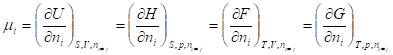
Где химический потенциал характеризует приращение соответствующего термодинамического потенциала при изменении количества данного вещества и постоянстве естественных переменных и количеств остальных веществ.
Если в системе совершается несколько видов работ, связанных с переносом вещества, то для описания равновесных состояний вместо химического потенциала используют понятие "полный потенциал", который помимо химического включает дополнительные вклады от этих видов работ.
Зная любой из потенциалов как функцию естественных переменных, можно с помощью основного уравнения термодинамики найти все другие термодинамические функции и параметры системы. Для этого используют соотношения Максвелла, в основе вывода которых лежит равенство смешанных производных характеристических функций:
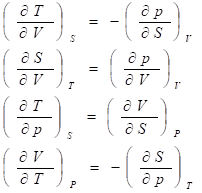
Характеристические функции как критерии направленности процессов
Согласно второму закону термодинамики при протекании самопроизвольных процессов в изолированных системах энтропия системы либо возрастает (необратимый процесс) либо остается неизменной (обратимый).
При протекании самопроизвольных процессов в закрытой системе при постоянстве объема и отсутствии полезной работы энергия Гельмгольца системы убывает в ходе необратимого процесса и остается неизменной при обратимом характере процесса.
То же самое можно сказать и об остальных характеристических функциях. Таким образом, условие возрастания энтропии в изолированной системе эквивалентно убыванию одного из термодинамических потенциалов (U, H, F, G) системы при фиксированных естественных переменных. В состоянии равновесия соответствующие термодинамические потенциалы достигают минимального значения.
Расчет изменения энергии Гиббса и энергии Гельмгольца в различных процессах
1) Изменение энергий Гиббса и Гельмгольца при изменении температуры
Зависимость энергии Гиббса и Гельмгольца от температуры в закрытых системах может быть определена с помощью фундаментальных уравнений:
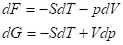
Функции F и G являются функциями состояния, для них:
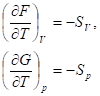
Для интегрирования этих уравнений надо знать температурную зависимость энтропии, которая определяется теплоемкостью системы. График зависимости энергии Гиббса от температуры в предположении линейной температурной зависимости теплоемкости приведен на рисунке.
2) Изменение энергий Гиббса и Гельмгольца при изотермическом расширении или сжатии.
Зависимость энергии Гиббса и Гельмгольца от давления и объема при постоянной температуре может быть определена интегрированием производных:
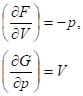
Для восстановления вида этой зависимости необходимо знать уравнение состояния фазы. Так, для идеального газа
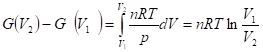
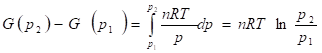
Если p1 = p = 1 бар, то говорят о стандартной энергии Гиббса, G°
3) Изменение энергий Гиббса и Гельмгольца при химической реакции.
Расчет изменения функций F и G в химических реакциях можно проводить разными способами. Рассмотрим три из них на примере энергии Гиббса.
I. По определению, G = H - TS. Если продукты реакции и исходные вещества находятся при одинаковой температуре, то изменение энергии Гиббса в химической реакции
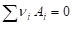
Равно
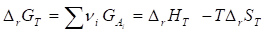
В стандартных условиях
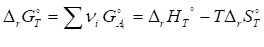
II. Аналогично тепловому эффекту реакции, изменение энергии Гиббса можно рассчитать, используя стандартные энергии Гиббса образования реагентов и продуктов:
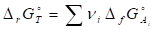
Или
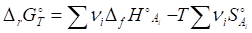
Изменение энергии Гельмгольца химической реакции между идеальными газами связано с энергией Гиббса
Где Дн - изменение количества молей газообразных веществ в ходе реакции. Для реакций в конденсированной фазе при небольших давлениях
III. Стандартная энергия Гиббса реакции может быть рассчитана с помощью стандартных приведенных потенциалов:
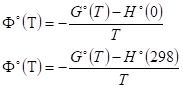
Где G°(T) - стандартное значение энергии Гиббса при температуре Т, H°(0), H°(298) - стандартные значения энтальпии при температурах 0 и 298 К. Функции Ф°(T) и Ф?°(T) вычисляются для газов по молекулярным данным, а для конденсированных фаз - на основании экспериментальных данных по теплоемкости. Связь между приведенными потенциалами и стандартной энергией Гиббса реакции выражается соотношениями:
Где ДrH°(0), ДrH°(298) - стандартные энтальпии реакции при 0 и 298 К.
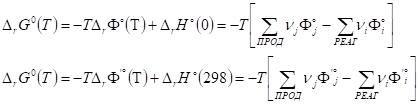
3. Растворы и их термодинамические свойства
Классификация растворов. Парциальные молярные величины. Идеальные растворы. Термодинамические свойства идеальных растворов. Химический потенциал компонента идеального раствора.
Равновесие "идеальный раствор Ї пар". Закон Рауля, его термодинамическое обоснование.
Неидеальные растворы. Активность и коэффициент активности. Стандартные состояния компонентов раствора.
Предельно разбавленные растворы. Законы Рауля и Генри для термодинамического описания свойств предельно разбавленных растворов.
Зависимость давления насыщенного пара от состава жидкого раствора. Уравнение Дюгема - Маргулиса и следствия из него. Положительные и отрицательные отклонения от закона Рауля. Законы Коновалова. Диаграммы состояния жидкость - пар. Перегонка и ректификация. Эбулиоскопия, криоскопия, осмотическое давление.
Общие сведения о растворах
Раствор - гомогенная система, состоящая из нескольких компонентов, т. е. образованная двумя и более индивидуальными веществами. Причем компоненты вступают в такое физико-химическое взаимодействие, что при изменении их соотношения в системе не происходит резкого изменения свойств раствора. Если компонентов два, то раствор бинарный (двойной или двухкомпонентный).
Растворы могут иметь различное агрегатное состояние. Кроме жидких растворов бывают также растворы газообразные и твердые. Газообразными растворами можно считать смесь газов и растворы жидкостей в газах. Твердые растворы получаются либо после охлаждения некоторых расплавов веществ, либо при растворении газов в твердых веществах.
В реальности - практически любая система является раствором, так как безпримесные системы и материалы - исключительная редкость.
Наука о растворах - одна из наиболее старых областей естествознания. По мере изучения свойств растворов были сформированы теории растворов. Одна из них - физическая (труды Вант-Гоффа, Аррениуса, Оствальда). Она предполагала, что в растворах молекулы растворенного вещества подобны молекулам идеального газа, они не взаимодействуют между собой и не взаимодействуют с растворителем. Другая теория растворов - химическая (труды Менделеева, Каблукова) указывала, что растворы нельзя считать простой механической смесью веществ, так как между всеми компонентами раствора всегда имеет место взаимодействие.
Вещества, которые могут быть выделены из раствора и существовать вне его, называются компонентами раствора. С термодинамической точки зрения деление их на растворитель и растворенные вещества условно. Обычно растворителем называют компонент, который либо присутствует в растворе в значительно большем количестве по сравнению с другими компонентами, либо имеет в чистом виде такое же агрегатное состояние, как и раствор (если остальные компоненты имеют при этом в чистом виде другое состояние). Принято то свойство, которое относится к растворителю, обозначать нижним индексом 1, а к растворенному веществу - либо индексом 2, либо буквой s.
Состав раствора
Понятие состава раствора имеет две стороны: качественную и количественную. Качественная характеристика состава раствора - это указание числа и природы (вида) компонентов, образующих раствор. Количественная характеристика состава раствора показывает число молей или массу каждого компонента, т. е. она является продолжением качественной характеристики. Числа молей и массы компонентов относятся к экстенсивным свойствам системы, в то время как давление и температура - к интенсивным. Чтобы оценить вклад компонентов раствора в интенсивные свойства, вводят на базе чисел молей или масс иные характеристики состава системы - концентрации.
Применяют различные способы выражения концентрации. Приведем в этом пособии только те, которые будем в дальнейшем использовать.
1. Мольные доли (или во многих учебниках - молярные доли).
Мольная доля компонента k () находится так:
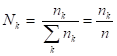
.
Принято какое-либо полное экстенсивное свойство раствора обозначать соответствующей буквой без индекса. В формуле (1) n - число молей всего раствора или общее число молей.
Очевидно, что
.
2. Массовые доли компонентов :
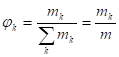
.
3. Молярная концентрация (с) - отношение количества вещества в молях к объему системы (моль/м3). Иногда эту концентрацию называют также плотностью числа молей [6], а саму концентрационную шкалу молярной концентрации - с-шкалой.
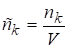
.
4. Массовая концентрация - это отношение массы компонента к объему системы (кг/м3).
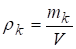
.
5. Моляльная концентрация раствора - это отношение количества растворенного вещества к массе растворителя (моль/кг). Эту концентрацию также можно назвать мольно-массовым отношением или концентрацией в d-шкале.
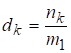
.
Концентрацию одного и того же раствора можно выразить различными способами, так как все виды концентраций связаны между собой.
Парциальное мольное свойство компонента раствора
Полное экстенсивное свойство чистого вещества k - , это же свойство, выраженное относительно одного моля вещества - .
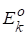
.
Свойства каждого компонента в растворе не будут равны свойствам этого компонента в виде чистого вещества, поэтому для описания свойств компонентов раствора используют понятие парциальных мольных свойств компонента k, которые обозначают.
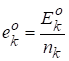
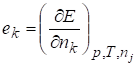
Записав это выражение для функции Гиббса, получим широко используемое в дальнейшем соотношение
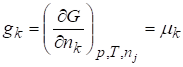
,
Где - парциальная мольная функция Гиббса;
- химический потенциал компонента k раствора, совпадающий с парциальной мольной функцией Гиббса.
Парциальное мольное свойство компонента раствора связано с мольным свойством раствора и полным свойством E всего раствора при помощи следующих уравнений:
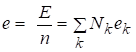
,
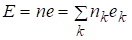
.
Идеальные и реальные растворы
В теории растворов рассматриваются два основных класса: идеальные и неидеальные (реальные) растворы. В идеальных растворах внутренняя энергия каждого компонента не зависит от концентрации, а парциальный мольный объем не изменяется при растворении. Идеальным считается такой раствор, в котором для каждого компонента при всех значениях давления (p), температуры (T) и мольной доли (NK) справедливо выражение зависимости химического потенциала компонента в этом растворе следующего вида:
,
Где - стандартный химический потенциал компонента k раствора, являющийся функцией давления и температуры. Стандартный химический потенциал компонента k - это потенциал при мольной доле этого компонента равной единице.
Стандартное состояние может быть реальным и гипотетическим. В зависимости от того, какое состояние компонента принимают за его стандартное состояние, различают три модели идеальных растворов: идеальный газовый раствор, идеальный совершенный раствор и идеальный предельно - разбавленный раствор.
В идеальном газовом растворе за стандартное состояние принято состояние компонента k в виде чистого идеального газа
.
Зависимость определяется уравнением для чистого идеального газа вида k:
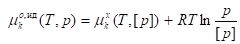
,
Где - стандартный химический потенциал чистого идеального газа k;
P - давление газа;
[p] - единица размерности давления.
Рассматривая совместно последние уравнения, получаем:
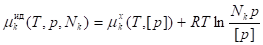
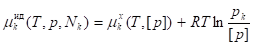
, ,
Где - парциальное давление компонента k в идеальном газовом растворе.
В идеальном совершенном растворе стандартное состояние любого компонента k - его состояние в виде чистого вещества.
.
Выражение для химического потенциала компонента k в этом растворе будет иметь следующий вид:
.
Идеальные совершенные растворы - это растворы, в которых и растворитель, и растворенные вещества обладают сходным химическим строением и имеют близкие термодинамические характеристики, например растворы оптических изомеров, или растворы изотопов.
Идеальный предельно разбавленный раствор - это раствор, в котором стандартные состояния для растворителя и растворенных веществ различны. Для растворителя за стандартное состояние здесь тоже принимают состояние в виде чистого вещества, а для растворенных веществ за стандартное состояние принимают их состояние в предельно - разбавленном растворе:
.
Причем:
.
Зависимость химического потенциала компонента реального раствора от концентрации можно записать в виде
,
Где - стандартный химический потенциал компонента k раствора;
- активность компонента k в растворе, определяемая как
- коэффициент активности.
Зависимость химического потенциала компонента k реального раствора от состава раствора в настоящее время устанавливают путем сравнения свойств компонента в реальном растворе с его свойствами в каком-либо идеальном растворе такой же концентрации. Тип идеального раствора выбирают по сходству реального раствора с идеальным: если это смесь газов, то разумно использовать идеальный газовый раствор; если это жидкий раствор с очень малой концентрацией растворенного вещества в растворителе, то удобно использовать идеальный предельно - разбавленный раствор и т. д.
Следовательно, имеются три системы сравнения (по числу типов идеальных растворов): на основе идеального газового раствора, идеального совершенного раствора, идеального предельно - разбавленного раствора. Две первые называются симметричными системами сравнения, последняя - несимметричной. Имеется различие между этими системами сравнения. Если остановиться на этом кратко, то в симметричной системе коэффициенты активности компонентов становятся равными единице в каждом из чистых компонентов (или в чистом идеальном газе). В несимметричной системе сравнения условия приближения коэффициентов активности к единице различны: для растворителя при, а для растворенного вещества при. Из одной системы сравнения нетрудно переходить в другую систему сравнения. Обычно принято указывать, в какой системе приведены коэффициенты активности: в симметричной системе сравнения на основе идеального совершенного раствора - ; на основе идеального газового раствора - и т. д.
Запишем выражения для химического потенциала компонента k неидеального раствора в симметричной системе сравнения:
Относительно идеального совершенного раствора
,
И относительно идеального газового раствора
,
Последнее уравнение с учетом (18) может также быть представлено как
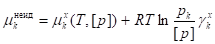
.
Руководствуясь общими соотношениями между термодинамическими функциями можно выразить важнейшие уравнения состояния, характеризующие реальные растворы.
Например, для симметричной системы сравнения на основе идеального газового раствора выражения для парциальных мольных энтропии, объема и энтальпии компонента k неидеального раствора будут следующими:
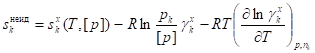
, х,
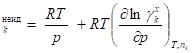
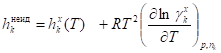
.
Функции смешения
Изменение экстенсивных свойств при образовании растворов удобно оценивать, используя специальные функции - функции смешения. Функция смешения - это изменение термодинамических функций Е при образовании раствора из чистых компонентов. Принято рассматривать процесс смешения, когда все компоненты находятся в одинаковом агрегатном состоянии, при одной и той же температуре и одинаковом давлении.
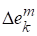
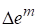
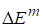
Для функций смешения используются оператор и верхний индекс m, например парциальные мольные функции смешения -, мольные функции смешения - , полные (или интегральные) функции смешения - . Между ними существуют те же соотношения, что и между парциальными мольными свойствами компонентов, мольными и полными свойствами раствора.
Парциальная мольная функция смешения, записанная для любого экстенсивного свойства E:
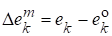
,
Где - парциальная мольная функция смешения компонента k;
- парциальное мольное свойство компонента k в растворе;
- мольное свойство чистого вещества k.
Мольная функция смешения:
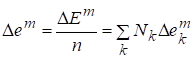
.
Полная функция смешения:
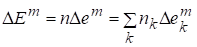
.
Основные функции смешения
|
Название функции смешения |
Выражение |
|
Химический потенциал смешения (или парциальной мольной функции Гиббса смешения). |
. |
|
Энтропия смешения | |
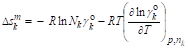
Объем смешения |
Х, |
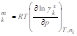
Энтальпия смешения |
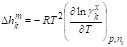
Если реальный раствор близок к идеальному, т. е. раствор можно считать идеальным совершенным, в котором равен единице, то:
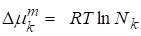
,
,
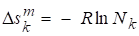
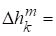
Х= 0, 0.
Таким образом образование идеальных растворов происходит без тепловых эффектов и изменения объема (расширения или сжатия). Процесс является самопроизвольным так как изменение энергии Гиббса при образовании идеального раствора отрицательно. Действительно мольная доля компонента k изменяется при образовании раствора, уменьшаясь от единицы до какого-то значения. Значит, логарифм будет отрицательный и функция Гиббса смешения тоже отрицательна. А в условиях постоянства температуры и давления именно функция Гиббса, а точнее, ее изменение является критерием самопроизвольности протекания процесса.
Мольная функция Гиббса смешения идеального раствора может быть найдена:
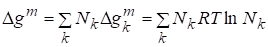
.
Полная функция Гиббса смешения рассчитывается по формуле:
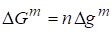
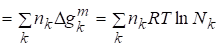
.
Избыточные термодинамические функции
Разность между экстенсивным свойством неидеального (реального) раствора с таким же свойством идеального раствора называют избыточной функцией. Избыточную функцию принято обозначать соответствующим символом экстенсивного свойства с верхним индексом Е - от англ. Еxcess - избыток).
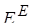
Существуют парциальная мольная избыточная термодинамическая функция (), мольная избыточная функция (), полная избыточная функция (). Между ними справедливы соотношения, аналогичные приведенным для парциальных мольных величин или для функций смешения.
.
Мольная избыточная функция будет равна
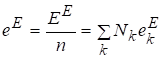
.
Полная избыточная функция найдется из соотношения
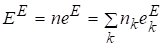
.
С учетом введенных обозначений можно записать:
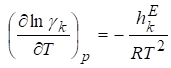
,
Х,
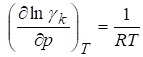
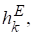
Где х - избыточная парциальная мольная энтальпия и избыточный парциальный мольный объем компонента k раствора.
Приведем далее некоторые избыточные функции для неидеального раствора в симметричной системе сравнения на основе идеального совершенного раствора.
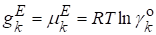
.
Мольная избыточная функция Гиббса:
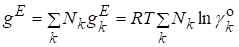
.
Полная избыточная функция Гиббса:
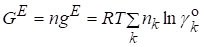
.
В соответствии со знаком функции говорят о положительном или отрицательном отклонении реального раствора от идеального поведения.
Парциальная мольная избыточная энтропия
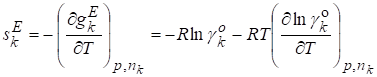
.
Парциальный мольный избыточный объем :
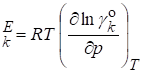
Х.
Парциальная мольная избыточная энтальпия:
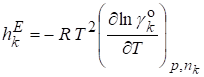
.
Избыточные термодинамические функции связаны с экспериментально измеряемыми на опыте величинами. Избыточная функция Гиббса связана с давлением пара над раствором. Избыточная энтальпия - это теплота смешения реального раствора при постоянном давлении, отнесенная к 1 молю раствора.
Термодинамические закономерности равновесий жидкость-пар в бинарных растворах. Уравнение Дюгема-Маргулеса
Парциальные давления веществ в паре, сосуществующем с жидкостью
При испарении бинарного раствора между жидкостью и паром устанавливается равновесие, представляющее собой частный случай равновесия в двухфазной двухкомпонентной системе. При небольших давлениях парообразную фазу можно рассматривать как смесь идеальных газов, в которой каждый компонент обладает определенным парциальным давлением. Однако парциальное давление компонента в паре определяется (наряду с общим давлением и температурой) концентрацией данного компонента в растворе. Поскольку для компонента идеальной газовой смеси:
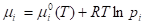
то:
Связь между изменениями парциальных давлений обоих компонентов раствора дает соотношение, которое может быть получено из уравнения Ван-дер-Ваальса. Путем ряда преобразований и с учетом того, что молярный объем пара намного превосходит молярный объем жидкости (так, при 0°С 1 моль идеального газа занимает объем 22,4 дм3, а объем 1 моль жидкости измеряется миллилитрами; например, 1 моль воды занимает 18 см3); основываясь на законе Дальтона было получено так называемое уравнение Дюгема-Маргулеса, которое может быть применено для проверки опытных данных о парциальных давлениях компонентов в паре над раствором.
Законы Коновалова
Связь между изменениями состава равновесных фаз и изменениями давления и температуры была впервые рассмотрена в работах Коновалова. Он исследовал экспериментально зависимость давления насыщенного пара над раствором от состава раствора. Обобщающие заключения сделаны им на основе результатов опытов и логических рассуждений, базирующихся на условиях устойчивости.
В настоящее время вывод законов Коновалова обычно проводится с использованием термодинамических закономерностей.
Первый закон Коновалова
Давление пара над раствором возрастает при увеличении в жидкости концентрации того компонента, содержание которого в паре больше, чем в растворе;
Давление пара над раствором уменьшается при увеличении в жидкости концентрации того компонента, содержание которого в паре меньше, чем в растворе;
Температура кипения раствора уменьшается при увеличении в жидкости концентрации того компонента, содержание которого в паре больше, чем в растворе;
Температура кипения раствора возрастает при увеличении в жидкости концентрации того компонента, содержание которого в паре меньше, чем в растворе.
Ход рассуждений, приведших к этим выводам, показывает, что закон справедлив для тех случаев фазовых равновесий, когда одна из фаз газообразна (испарение, возгонка), так как лишь в этом случае можно утверждать, что дифференциальные объемные эффекты и дифференциальные теплоты образования одной фазы из другой положительны. Но и для испарения закон Коновалова может быть применен лишь при температурах и давлениях, достаточно далеких от критических, ибо вблизи критической точки, где свойства пара и жидкости одинаковы, знаки объемного эффекта и дифференциальной теплоты испарения неопределенны.
Отклонения от закона Рауля нередко приводят к возникновению минимумов (максимумов) на кривых зависимости давления пара (температуры кипения) от состава фаз. Свойства систем, обладающих экстремумами температуры или давления,
Определяются с помощью второго закона Коновалова.
Второй закон Коновалова можно сформулировать так:
В точках экстремума давления пара при изотермических условиях или температуры кипения при изобарических условиях составы пара и жидкости одинаковы. В отличие от первого второй закон Коновалова справедлив, при любых (в том числе и близких к критическим) температурах и давлениях и для любых случаев фазовых равновесий в двухфазных системах.
Системы, имеющие азеотропы, широко распространены. Азеотропными (нераздельнокипящими) называются растворы, обладающие экстремумами давления и температуры,
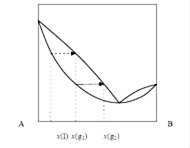
При этом наиболее часто встречаются растворы, обладающие максимумом давления и минимумом, температуры кипения. К их числу относятся системы: вода -- этанол, метанол -- ацетон, бензол -- уксусная кислота, бензол -- циклогексан, метил-циклогексан -- толуол и т. д. Азеотропов с минимумом давления и максимумом температуры кипения известно значительно меньше. К их числу относятся: вода -- соляная кислота, вода -- серная кислота, вода -- муравьиная кислота, хлороформ -- ацетон и т. д.
Третий закон Коновалова характеризует влияние изменения состава одной из фаз на состав другой фазы. Третий закон Коновалова имеет те же ограничения, что и первый. В случае систем раствор -- пар, он теряет свою силу для состояний близких к критическому.
Похожие статьи
-
Основы химической термодинамики. Первое начало термодинамики Термодинамические системы и термодинамические параметры. Функции состояния. Парциальные...
-
ХИМИЧЕСКАЯ ТЕРМОДИНАМИКА - Неограническая химия
Задание 3.1. Запишите реакцию взаимодействия указанного по варианту элемента с кислородом. Используя приведенные в табл. III.1 данные, рассчитайте...
-
Второе начало термодинамики - Физическая химия
Самопроизвольные и несамопроизвольные процессы. Термодинамически обратимые и необратимые процессы. Работа и теплота обратимого процесса. Формулировка...
-
Растворы - термодинамические устойчивые системы переменного состава, состоят не менее чем из двух компонентов и продуктов их взаимодействия. Это...
-
Физические свойства Таллия - Третья группа периодической системы
. Таллий мягкий металл, на воздухе легко окисляется и быстро тускнеет. Таллий при давлении 0,1 Мн/м2 (1 кгс/см2) и температуре ниже 233 °С имеет...
-
Кремний - элемент главной подгруппы четвертой группы третьего периода периодической системы химических элементов Д. И. Менделеева, с атомным номером 14....
-
Физические свойства молока - Химия и физика молока
1). Плотность, вязкость, поверхностное натяжение. 2). Осмотическое давление и температура замерзания. 3). Удельная электропроводность. Плотность молока...
-
А) Углерод (С), кремний (Si), германий (Ge), олово (Sn), свинец (РЬ) - элементы 4 группы главной подгруппы ПСЭ. На внешнем электронном слое атомы этих...
-
Физические свойства., Химические свойства. - Третья группа периодической системы
Алюминий в свободном виде -- серебристо-белый металл, обладающий высокой тепло - и электропроводностью. Алюминий имеет невысокую плотность -- примерно...
-
В термодинамике понятие "энтропия" было введено Р. Клаузиусом (1865), который показал, что процесс превращения теплоты в работу следует общей физической...
-
Основные понятия химической термодинамики - Химическая термодинамика. Термохимия. Решение задач
Прежде чем приступить к изучению предмета химической термодинамики, необходимо ввести ряд терминов и понятий, используемых в этом разделе. Изучаемые...
-
Введение в физическую химию - Физическая химия
Физический химия термодинамика Предмет и задачи физической химии. Методы исследования в физической химии: термодинамический, статистический,...
-
Алюминий - основной представитель металлов главной подгруппы III группы Периодической системы. Свойства его аналогов - галлия, индия и таллия -...
-
Внутренняя энергия термодинамическая функцция состояния системы, ее энергия, определяемая внутренним состоянием. Внутренняя энергия складывается в...
-
Энтропия. Движущее начало химических процессов - Химическая термодинамика. Термохимия. Решение задач
Убедившись в полезности знания тепловых эффектов химических превращений, мы, тем не менее, не смогли ответить на вопрос: "Почему одни химические реакции...
-
Термодинамика - наука о взаимопревращениях различных форм энергии и законах этих превращений. Термодинамика базируется только на экспериментально...
-
Первое начало термодинамики. Энтальпия - Химическая кинетика, равновесия, термодинамика
Это закон сохранения энергии. Подводимая к системе теплота Q расходуется на изменение ее внутренней энергии и на совершение работы А: Q = U + А Здесь U =...
-
Допустим, реакция aA + bB <> хХ + yY - гомогенная и идет в одну стадию. Тогда скорость прямой реакции vПр = KПр [A]A{B]B, а скорость обратной...
-
Положение металлов в ПС. Физические свойства металлов. Методы получения металлов - Основы химии
Металлы располагаются в основном в левой и нижней части ПС К физическим свойствам относятся плотность, плавление (температура плавления),...
-
Ниже рассматривается наиболее важный изобарно-изотермический случай. Если реакция идет в изохорно-изотермических условиях, то вместо энергии Гиббса нужно...
-
Фактически это следствие первого начала термодинамики, но сформулирован раньше, чем первое начало. Тепловой эффект изобарного (или изохорного) процесса...
-
Свойства растворов неэлектролитов. Замерзание и кипение растворов. Законы Рауля - Основы химии
По наличию или отсутствию электрической проводимости растворы веществ делят на электролиты - проводящие электрический ток, и неэлектролиты - не...
-
Если на равновесную с-му не оказ-ся вноешнего воздействия (не изм. темп, давл.), то равновесие м/существовать неизменным долго. Любое внешнее возд-ие...
-
В предыдущих разделах рассмотрены отдельные аспекты кинетики, а теперь пора подвести общие итоги: от чего зависят скорости реакций и как можно ими...
-
Химическим равновесием называется такое состояние химической системы, при котором количества исходных веществ и продуктов не меняются со временем. 1....
-
Критерии самопроизвольного протекания процессов - Химическая кинетика, равновесия, термодинамика
А) В изолированной системе самопроизвольно идут только процессы с увеличением энтропии. S > 0 - процесс возможен, S Одновременно действуют обе тенденции...
-
К числу физических факторов, вызывающих коррозию цементного камня и бетона, относят их попеременное увлажнение и высыхание, которое сопровождается...
-
Основные показатели качества природных вод - Химия воды
Температура -- физический показатель качества воды. Температура воды поверхностных водоисточников в зависимости от сезона года изменяется от 0 до 30°С....
-
Энтропия. Самопроизвольный процесс - Химическая термодинамика и ее процессы
Самопроизвольный процесс - процесс, который может протекать без затраты работы извне, причем в результате может быть получена работа в количестве,...
-
Скорость реакции определяется изменением молярной концентрации одного из реагирующих веществ: V=dC/dtV. Факторы, влияющие на скорость химических...
-
Общие сведения Термодинамика - наука о превращениях энергии в различных процессах, как физических, так и химических, и о направлении процессов, о...
-
1. Какие вопросы решает химическая термодинамика? 2. Что называется термодинамической системой? Как классифицируются системы? Приведите примеры различных...
-
Понятие квантовой химии - Квантовые концепции в химии
Квантовая химия - это раздел теоретической химии, в котором строение и свойства химических соединений, их взаимодействие и превращение в химических...
-
(а) Зависят ли энергия активации и предэкспоненциальный множитель от природы реагентов? от их концентраций? от температуры? от присутствия посторонних...
-
Краткая теория В старом учебнике Глинки вопрос хорошо изложен на качественном уровне, но не хватает строго количественного описания влияния температуры...
-
Жидкостное восстановление и радиолиз - Коллоидная химия
Энергия наночастица жидкостный радиолиз Жидкофазное восстановление. Жидкофазное восстановление. Химические восстановление зависит как от природы пары...
-
Специфические способы определения удельной свободной поверхностной энергии - Коллоидная химия
Поверхностная энергия , энергия, сосредоточенная на границе раздела фаз, избыточная по сравнению с энергией в объеме. При увеличении поверхности раздела...
-
Смещение химического равновесия. Принцип Ле-Шателье - Основы химии
Положение химического равновесия зависит от следующих параметров реакции: температуры, давления и концентрации. Влияние, которое оказывают эти факторы на...
-
Физические и химические свойства простого вещества - Свойства германия
Начать следует с описания физических свойств простого вещества германия. Германий представляет собой твердый серебристо-белый полуметалл, обладающий...
-
Применение H2O2 связано с его окислительными свойствами и безвредностью продукта его восстановления (H2O). Его использую для отбеливания тканей и мехов,...
Третье начало термодинамики - Физическая химия