Применение газовой хроматографии для исследований адсорбции и определения удельной поверхности твердых тел - Определение теплоты и энтропии адсорбции или растворения на основе хроматографических данных
Газовый хроматография энтропия адсорбция
Области физико-химических применений газовой хроматографии
Газовая хроматография, имеющая главным образом широкое аналитическое применение, в настоящее время находит также все большее применение и в физической химии для быстрого исследования различных явлений в газах, растворах и на поверхности твердых тел и определения соответствующих физико-химических констант. Сюда относятся применения газохроматографического метода для определения коэффициентов активности растворов, энтропии и теплот растворения, давления паров, определение молекулярных весов, коэффициентов диффузии газов, изотерм адсорбции, теплот и энтропии адсорбции, исследование межмолекулярного взаимодействия адсорбат - адсорбент и адсорбат - адсорбат, а также удельной поверхности твердых тел, кинетики адсорбции, кинетики каталитических реакций, констант равновесия различных реакций, энергии активации внутренней диффузии, температур кипения высококипящих углеводородов, измерения межфазного сопротивления газ-жидкость.
Газохроматографический метод определения этих физико-химических характеристик выгодно отличается от соответствующих статических методов быстротой измерения и возможностью применения простой стандартной невакуумной аппаратуры (причем часто при сохранении и в ряде случаев даже при значительном увеличении точности измерения), а также возможностью работы в широких интервалах температур, что позволяет исследовать многие растворяющиеся или адсорбирующиеся вещества. Газохроматографическим методом в отличие, например, от калориметрического легко проводить адсорбционные исследования при очень малых заполнениях поверхности и при значительно более высоких температурах. Кроме того, газохроматографическим способом можно измерить изотермы адсорбции агрессивных веществ (в частности, содержащих фтор, серу и т. п.). В аппаратуре для статических опытов такие измерения затруднительны.
Вычисление равновесной изотермы адсорбции из "равновесной" хроматограммы
Количественная связь изотермы равновесия (распределения) между подвижной и неподвижной фазами и "равновесной" хроматограммон впервые исследована для варианта хроматографии жидкость - поверхность твердого тела Вильсоном. Эти исследования продолжены Вейсом и де Во и в основном Глюкауфом. Глюкауф разработал метод расчета изотермы равновесия из растянутой границы выходной кривой, допустив соблюдение условий равновесной хроматографии (т. е. мгновенную диффузию и мгновенное установление адсорбционного равновесия в колонке). Им экспериментально доказана справедливость такого расчета для хроматографии жидкость - твердое тело.
Викке, Кремер, Джемсом и Филлипсом этим способом впервые определили изотермы адсорбции из газохроматографических измерений. Грегг и Сток применили этот метод для расчета изотерм адсорбции паров в широком интервале давлений и для различных форм выходных кривых, соответствующих различным формам изотерм адсорбции.
Метод Глюкауфа очень прост и позволяет быстро получить изотермы адсорбции из хроматограмм. Недостатки этого метода связаны с допущением о мгновенном установлении равновесия, т. е. в нем не учитывается диффузионное размывание и размывание, связанное с задержкой массообмена между подвижной и неподвижной фазами. Для уменьшения диффузионного размывания Веспалек и Грубнер применили короткие колонки, наполненные мелкозернистым адсорбентом (порошком), что приближает условия работы колонки к равновесным. В работах Шая и Рогинского с сотр. использовался метод Глюкауфа и приведены некоторые указания, облегчающие практические расчеты изотерм адсорбции из газохроматографических данных.
В теории равновесной хроматографии предполагается, что в соответствующих газохроматографических опытах соблюдены условия, практически устраняющие диффузионное и кинетическое размывание хроматографической полосы. Поэтому искажение этой полосы связывается только с отклонением равновесной изотермы адсорбции от изотермы Генри. Такой равновесной, но не идеальной хроматографии отвечает дифференциальное уравнение материального баланса в элементарном слое колонки (равенство скорости подачи адсорбата с газом-носителем и скорости распределения его между газовой фазой и адсорбционным слоем на поверхности адсорбента):
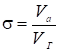
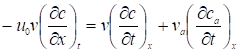
(1)
Где u0 - линейная скорость газа-носителя; v - объем газовой фазы в колонке; va - объем адсорбированного слоя; с - концентрация адсорбата в газовой фазе, cа - концентрация адсорбата в неподвижном (адсорбционном) слое на поверхности адсорбента; t - время, прошедшее от ввода пробы; х - расстояние от начала колонки (со стороны входа газа) до рассматриваемого элементарного слоя. Это уравнение легко преобразуется в уравнение
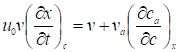
, (2)
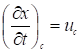
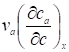
Где линейная скорость перемещения вдоль колонки концентрации с, a в условиях равновесной хроматографии представляет собой наклон изотермы адсорбции в точке, соответствующей концентрации с, так как в этом случае
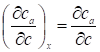
, a -
Соответствующее изменение величины адсорбции а (m - масса адсорбента в колонке). Таким образом, из уравнения (IV,2) следует, что
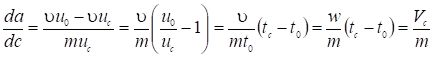
, (3)
Где w - объемная скорость неадсорбирующегося газа-носителя; tс и t0 - времена удерживания соответственно адсорбата при его концентрации с в газовой фазе и газа-носителя, a Vc - удерживаемый объем адсорбата при концентрации с (при температуре колонки). Отсюда величина адсорбции при равновесной концентрации с в газовой фазе
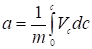
. (4)
Уравнение (4) позволяет найти величину а для разных значений равновесной концентрации с, т. е. изотерму адсорбции
А = ц (с)
Для численного расчета изотермы в это уравнение необходимо ввести значение удерживаемого объема Vc и концентрации с, выраженные через величины, записанные на диаграммной ленте в хроматографическом опыте.
Как для вычисления интеграла в уравнении (4), так и для определения равновесной концентрации адсорбата в газовой фазе с, необходимой для построения изотермы адсорбции а = ц (с) или а = (р), где р = cRT - парциальное давление адсорбата в газовой фазе, необходимо выразить показания детектора в единицах концентрации с. Так как отклонения пера самописца h обычно пропорциональны с, то
(5)
Где k - постоянная величина для данного адсорбата и данного диапазона чувствительности детектора. Величина k может быть определена фронтальным методом из определений отклонений пера самописца h при разных концентрациях с газа-носителя. Этот метод вполне надежен, но требует создания точно известных концентраций в газе-носителе. Кроме того, не все хроматографы приспособлены для работы фронтальным методом. Поэтому удобнее определить k из самих проявительных хроматограмм, вводя в колонку достаточно точно определенное количество адсорбата mа. После выхода из колонки
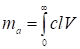
, (6)
Где V - объем протекающего через колонку газа, а пределы интегрирования соответствуют вводу пробы и полному ее вымыванию из колонки. Таким образом, учитывая (5), получаем:
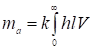
. (7)
Обычно на самописце дается не шкала V, а шкала длины диаграммной ленты z в направлении ее движения. Скорость движения диаграммной ленты равна:
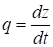
(8)
Так как объемная скорость газа-носителя в детекторе составляет:

, (9)
То из выражений (8) и (9) следует, что
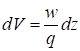
. (10)
Подставляя это выражение в уравнение (7) получаем, что
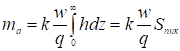
(11)
Где площадь пика
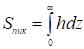
(12)
И представляет собой площадь под всей выходной кривой проявительной хроматограммы адсорбата. Таким образом, калибровочная константа детектора равна:
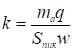
. (13)
Подставляя далее в формулу (4) выражения dc = kdh и
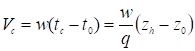
,
Где zh-z0 - расстояние на диаграммной ленте самописца от момента выхода неадсорбирующегося компонента до момента выхода газа с концентрацией с адсорбата (т. е. до соответствующего отклонения h), получаем, что
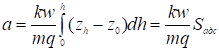
, (14)
Где
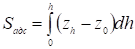
(15)
Представляет собой площадь, определяемую на диаграммной ленте самописца между осью h при z = z0 и растянутым краем пика адсорбата. На рис. 1 показаны примеры определения этой площади для двух типов пиков с растянутым задним и с растянутым передним краем. Эти пики соответствуют не имеющим перегиба изотермам адсорбции, обращенным выпуклостью к оси адсорбции и соответственно к оси концентрации (или давления газа).
В формуле (13) величина w представляет собой скорость потока газа в детекторе при температуре колонки. Подставляя выражение (13) для k в формулу (14), получаем, что величина адсорбции
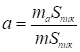
. (16)
Этой простой формулой обычно и пользуются для определения адсорбции из записанной на диаграммной ленте хроматограммы.
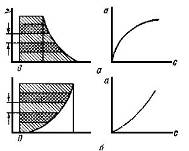
Рис. 11 Графическое интегрирование хроматографических кривых и вычисленные из них изотермы адсорбции. Слева на оси ординат - h, на оcu абсцисс - z. а - пик с острым фронтом и растянутой задней границей; б - пик с растянутым фронтом и острой задней границей
При этом необходимо, однако, принимать во внимание сделанное предположение о постоянстве скорости потока газа в детекторе w и скорости диаграммной ленты самописца q.
Следует учесть также зависимость w от перепада давления в колонке и поправки, связанные с отличием давления и температуры в измерителе скорости потока газа от давления у выхода из колонки и от температуры колонки.
Соответственно из выражений (13) следует, что равновесная этой величине адсорбции а концентрация адсорбата в газе составляет:
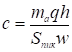
(17)
И его парциальное давление
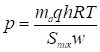
. (18)
Этой формулой пользуются для определения равновесного давления из записанной на диаграммной ленте самописца хроматограммы.
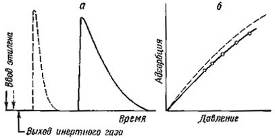
Рис. 12 Хроматографические пики этилена на активированном угле при 101С (а), из которых ычислены изотермы адсорбции (б). Сплошные кривые - колонка длиной 1 м с набивкой 6,91 г активного угля; пунктирные - колонка длиной 30 см набивкой 1,6 г активного угля; О - данные статических измерений
Таким образом, для получения точки на изотерме адсорбции а = (p) можно обойтись без отдельной калибровки детектора, если величина введенной пробы mа точно известна, величины w и q во время опыта сохранялись постоянными, а величины h, Sпик и Sадс найдены на хроматограмме.
Таким способом Хубер и Кейлеманс из проявительных хроматограмм с растянутым задним краем вычислили изотерму адсорбции этилена на угле (обращенную выпуклостью к оси а) при 100°С в удовлетворительном соответствии со статическими измерениями (рис. 2).
На рис. 3 приведены хроматограммы для адсорбции воды на однородной поверхности графитированной термической сажи при разных величинах проб и разных температурах, а на рис. 4 - соответствующие изотермы адсорбции. Поскольку в этом случае очень слабого неспецифического взаимодействия адсорбат - адсорбент и относительно сильного и специфического взаимодействия адсорбат - адсорбат изотерма адсорбции обращена выпуклостью к оси давления пара, хроматограммы имеют растянутый передний край. На рис. 3 указан способ интегрирования такой хроматограммы. Растянутые задние края пиков в области низких h сохраняются и в колонке без адсорбента. Поэтому интегрирование велось по растянутому переднему краю пиков. Точки на каждой изотерме соответствуют разным h на кривой переднего края хроматограммы, огибающей все пики (на рис. 3 эти кривые показаны жирными линиями).
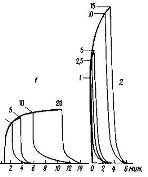
Рис. 13 Примеры хроматограмм воды на графитированной термической саже при различных величинах проб (указаны на рисунке в мкл) и температурах 32°С (1) и 50°С (2) (колонка 100x0,4 см, скорость газа-носителя - гелия50 мл/мин, детектор - катарометр хроматограф "Цвет-1")
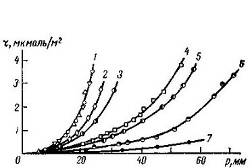
Рис. 14 Изотермы адсорбции пара воды на графитированной термической саже при разных температурах, вычисленные из хроматограмм
1 - 20°С, 2 - 32°С, 3 - 34°С; 4 - 40°С; 5 - 43°С, 6 - 60°С На изотерме 29°С разными значками обозначены точки, относящиеся к различным скоростям потока газа (от 40 до 60 мл/мин)
Представляет большой интерес определение из хроматограмм изотерм адсорбции с точками перегиба. Впервые это определение было сделано Греггом и Стоком фронтальным методом (для системы циклогексан, бензол - силикагель). Практически весьма удобно определять такие изотермы адсорбции из проявительных хроматограмм. Рассмотрим два важных примера - изотермы адсорбции с одной и двумя точками перегиба. На рис. 5 представлены хроматограммы бензола на одной поверхности графитированной термической сажи (сильное специфическое взаимодействие адсорбат - адсорбат).
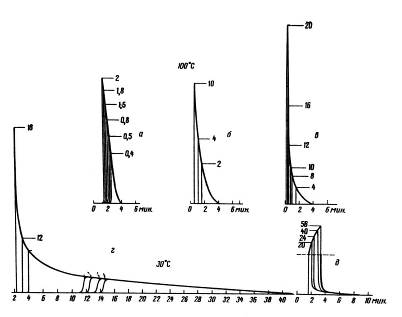
Рис. 15 Хроматограммы бензола на графитированной термической саже при разных величинах проб и температурах (условия опыта те же, что и на рис. 52)
Жирные линии - огибающие кривые, по которым велось интегрирование
В этом случае изотерма относится ко второму типу по Брунауэру (см. рис. 6) и довольно хорошо описывается уравнением Брунауэра, Эммета и Теллера (уравнение БЭТ. Из хроматограмм рис. 5 видно, что на разных высотах h пик имеет разную форму. При низких h растянут задний край хроматограммы (см. хроматограммы а - г на рис. 5). Это соответствует изотерме адсорбции, обращенной выпуклостью к оси адсорбции (когда адсорбция бензола происходит преимущественно в первом слое рис. 6).
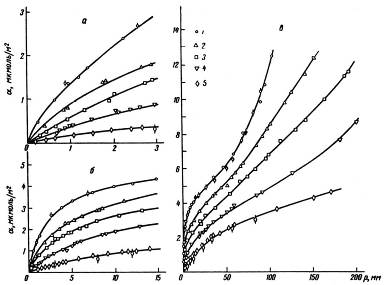
Рис. 16 Изотермы адсорбции бензола на графитированной термической саже при 30°С (7), 409С (2), 50°С (3), 70°С (4) и 100°С (5), вычисленные из хроматограмм, которые даны на рис. 54 а - область малых заполнений; б - средняя область; в - область больших заполнений. Перечеркнутые точки -- статические измерения
При высоких h растянут уже передний край хроматограммы (хроматограмма д на рис. 5,). Это соответствует той части изотермы адсорбции бензола на графитированной саже, которая после перегиба обращена выпуклостью к оси давления пара (когда адсорбция бензола происходит преимущественно во втором и последующих слоях). В средней части изотермы вблизи точки перегиба на изотерме адсорбции имеется участок, довольно близкий к линейному. В соответствии с этим на хроматограмме при увеличении пробы рост h происходит при практически постоянном значении z, т. е. оба края хроматограммы в этой средней области h практически вертикальны (хроматограммы д на рис. 5). На рис. 6 можно видеть эти три области изотермы адсорбции.
Следующий интересный пример - адсорбция пара метанола на однородной поверхности графитированной термической сажи. Взаимодействие адсорбат - адсорбент остается неспецифическим, но благодаря наличию в молекуле метанола метильной группы оно более сильно, чем при адсорбции воды, хотя и менее сильно, чем при адсорбции зола. Взаимодействие же адсорбат - адсорбат сильное и специфическое (в этом случае возможна ассоциация молекул метанола в адсорбционном слое с образованием взаимных водородных связей. Благодаря этому при малых давлениях пара изотерма адсорбции метанола на графитированной термической саже обращена выпуклостью к оси давления пара (рис. 7).
По мере роста поверхностной концентрации молекул метанола между ними все сильнее проявляются специфические взаимодействия, и изотерма начинает круто подниматься.
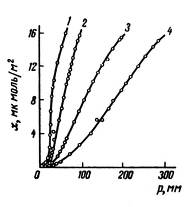
Рис. 17 Изотермы адсорбции пара метанола на графитированной термической саже при разных температурах, полученные из хроматограмм (рис. 57) 1 - 20°С; 2 -30°C; 3 -42°С; 4 -53°С. Точки для 20°С получены статическим методом
Однако ограниченность места на поверхности приводит к тому, что рост адсорбции в первом слое замедляется, в результате чего изотерма проходит первую точку перегиба. С дальнейшим увеличением давления пара начинает, однако, сказываться переход к адсорбции во втором и последующих слоях, что вызывает снова ускорение роста адсорбции, в результате чего изотерма адсорбции проходит вторую точку перегиба.
На рис. 8 представлены соответствующие хроматограммы, полученные при разных величинах проб и при разных температурах колонки. При низких температурах изотермы адсорбции (рис. 6) имеют два участка, обращенных выпуклостью к оси давлений, и один средний участок, обращенный вогнутостью к этой оси, причем вблизи и 0,5 такие изотермы имеют разрыв, т. е. одному значению р соответствует несколько значений а (термодинамически неустойчивые состояния).
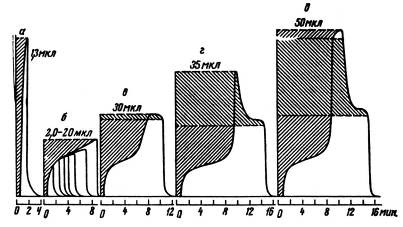
Рис. 18 Хроматограммы метанола на графитированной саже при 30
Соответственно этому при высоких температурах и более низких h хроматограммы имеют только один передний растянутый край, а хроматограммы при низких температурах и более высоких h имеют вначале растянутый передний край, затем, в области разрыва изотермы, большой участок, соответствующий нескольким значениям а при одном и том же значении р, далее при еще более высоких h - растянутый задний край и, наконец, при еще больших h (после второго перегиба изотермы) - растянутый передний край. Края хроматограмм, соответствующие устойчивым состояниям, на рис. 8 обозначены жирными линиями.
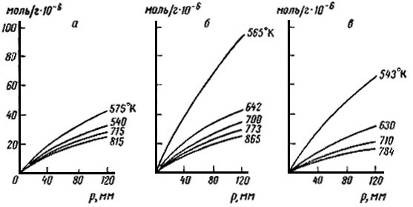
Рис. 19 Изотермы адсорбции гексана при различных температурах, измеренные газохроматографическим способом. а - на силикагеле, б - нп алюмогеле, в - на алюмосиликагеле
Из приведенных на рис. 2, 3, 5 и 8 хроматограмм видно, что при благоприятном выборе длины колонки, скорости газа, соотношения между размерами молекулы адсорбата и размерами пор и температуры диффузионные процессы и кинетика адсорбционно десорбционного обмена не искажают в заметной степени форму хроматографического пика, которая определяется, в основном, формой равновесной изотермы адсорбции. В пользу этого говорит совпадение растянутых краев пиков для разных величин проб и очень близкое к вертикальному положение острых краев пиков.
Следует отметить, что Киппинг и Уинтер при использовании метода газовой хроматографии для исследования изотерм адсорбции не рекомендуют вычислять изотерму адсорбции из одной выходной кривой, полученной методом проявительной газовой хроматографии, так как иногда задние фронты пиков (при некоторых типах изотерм) для разных величин проб не совпадают, в частности при адсорбции воды на кремнеземах (рис. 10).
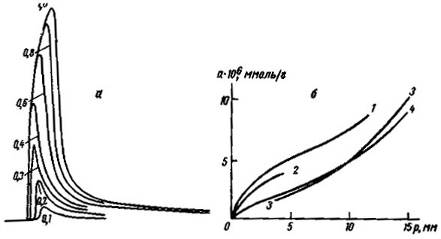
Рис. 20 Хроматограммы воды (величины проб указаны на рис. в мл) (а) и изотермы адсорбции воды на кливландите, вычисленные из проявительной хроматограммы с пробами 0,6 мл (1) и 0,3 мл (2), фронтальным методом (3) и по одной проявительной хроматограмме с максимальной пробой (4) (б)
Если задние границы пиков, полученных при разных дозах, не совпадают, газохроматографический метод не может дать хороших количественных результатов, но его можно использовать для качественного суждения об адсорбционных свойствах. Однако при получении хроматограммы не слишком сильно адсорбирующихся веществ при достаточно аккуратном вводе и измерении величины пробы и при выборе оптимальной скорости газа подобные затруднения обычно отпадают.
Биби, Эванс, Кляйнштубер и Ричардс вычислили изотермы адсорбции азота, аргона, кислорода, окиси углерода и гексафторэтилена на графитированной саже из хроматограмм, полученных фронтальным методом.
Как впервые было показано Грином и Пастом, газохрамографическим методом при соблюдении некоторых условий легко можно определить теплоты адсорбции. Если в хроматографическом опыте практически достигается равновесие и изотерма адсорбции начинается линейным участком (случай так называемой идеальной равновесной хроматографии), то ее наклон выражает константу Генри:
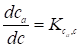
, (19)
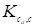
Где индексы у константы Генри указывают на то, что концентрации адсорбированной и газовой фаз выражены в одинаковых единицах. Из уравнения (3) в этом случае следует, что Vc (или VR) не зависит от концентрации (узкий симметричный пик) и
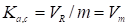
, (20)
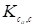
Где Vm - удельный удерживаемый объем, выраженный в см3 газа при температуре колонки к 1 г адсорбента, а индексы у константы Генри указывают на то, что адсорбция а отнесена к 1 г адсорбента, а содержание компонента в равновесной газовой фазе отнесено к единице ее объема (например, моль/л или ммоль/мл).
Для вычисления констант равновесия и далее теплоты адсорбции из симметричных пиков необходимо определить исправленные удерживаемые объемы по максимума узких симметричных пиков. Величины исправленных удерживаемых объемов VR - рассчитываются из хроматограмм по следующей формуле:
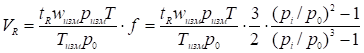
(21)
Где tR - исправленное время удерживания; wизм - объемная скорость газа-носителя в измерителе расхода газа;
Ризм и Тизм - давление и температура в измерителе расхода газа; Т - температура колонки; pi и р0 - давления газа соответственно у входа и у выхода из колонки.
Физико-химическими константами, не зависящими от величины удельной поверхности, являются следующие величины:
Ka, c = Ka, c/s=Vs, (22)
Где
Vs = Vm/s (23)
Представляет собой удерживаемый объем на единицу поверхности адсорбента (непористого или достаточно широкопористого).
Если из изотермы адсорбции а = ц (с) нужно перейти к изотерме адсорбции а = (р), то, принимая во внимание, что р = cRT, получаем:
Ka, p = Ka, c/RT=Vs/RT. (24)
Зная константу равновесия, можно легко найти стандартное изменение химического потенциала адсорбата м (дифференциальной свободной энергии) при адсорбции
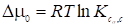
(25)
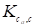
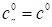
Где безразмерная константа равновесия определена через объемные концентрации в адсорбционном пространстве и в равновесном свободном объеме газа. За стандартные состояния в объеме газа и в адсорбционном объеме здесь приняты равные концентрации, соответствующие давлению в объеме газа в 1 атм. Концентрация в адсорбционном объеме равна:
, (26)
Где ф - толщина адсорбционного слоя. Поэтому, согласно уравнениям (20) и (22),
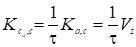
. (27)
Таким образом,
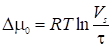
. (28)
Величину Vs в этом уравнении необходимо подставить в см3 (удерживаемый объем газа при температуре колонки Т) на единицу поверхности s (в см2), а ф - в см. В этом случае величина Vs/ф становится безразмерной.
Практически удобнее определять величину м0 по формуле
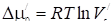
(29)
Подставляя величины в удобных по абсолютным значениям единицах - см3/м2. Связь между мc " и дается выражением
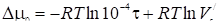
, (30)
Если ф выражена в см, a - в см3/м2, или
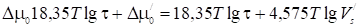
, (30a)
Если ф выражено в A, a - в см3/м2 (мл/м2).
Изостерическая теплота адсорбции (положительной условимся считать выделяющуюся теплоту) равна:
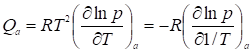
. (31)
В области Генри она не зависит от а:
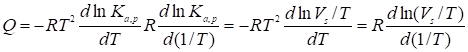
. (32)
Так как удельная поверхность s практически и масса m адсорбента вообще не зависят от Т, то величины Q могут быть определены непосредственно из зависимости от температуры экспериментальных величин удельных объемов удерживания Vm или объемов удерживания в данной колонке Vr:
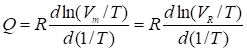
. (33)
Таким образом, в случае узких симметричных пиков можно легко получить изостерическую теплоту адсорбции Q, определяя Vs (или Vm, VR) при нескольких температурах колонки Т (в возможно более широком интервале Т) и строя график зависимости lg (Vs/T) (или lgVm/T, lg VR/T) от 1/T.
На рис. 11 (вверху) приведены хроматограммы ксенона на кристаллах цеолита NaX, полученные при разных температурах колонки. Эти пики достаточно узки и симметричны благодаря неспецифическому взаимодействию благородных газов с цеолитами и слабому взаимодействию адсорбат - адсорбат при малых заполнениях. На рис. 11 (внизу слева) приведена зависимость lg VR/T от 1/T, а на рис. 11 (внизу справа) зависимость lg VR/T от 1/T (величины Vr определены по максимумам пиков хроматограмм рис. 7, а).
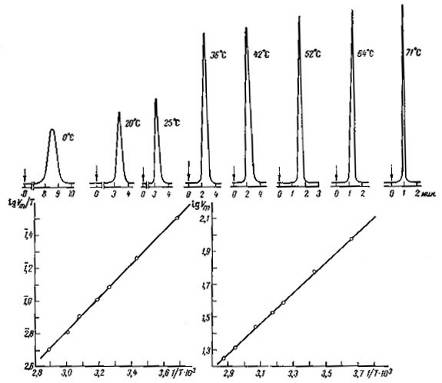
Рис. 21 Хроматограммы ксенона на кристаллах цеолита типа NaX (спрессованных в гранулы без связующего) при разных температурах колонки (колонка 40 х 0,4 см, газ-носитель - гелий, детектор - катарометр, хроматограф "Цвет-1") (а); соответствующая зависимость lgVm/ToT i/T (б), соответствующая зависимость lg Fm от i/T (в)
Из рис. 11 (внизу) видно, что эти зависимости являются практически линейными (теплота адсорбции на однородной поверхности при малых заполнениях несильно зависит от температуры). Из формул (32) и (33) следует, что умножение наклона графика зависимости lg (VS/T) или lg (Vm/T), lg (VR/T) от 1/T (рис. 11, внизу слева) на 2,303 R = 4,575 дает теплоту адсорбции Q в кал/моль. Для определения Q из графика зависимости lg Vs (или lg Vm, lg Vr) от 1/T (см. рис. 11, внизу справа) следует умножить наклон на 4,575 и прибавить RT = 1,987T кал/моль (для средней температуры интервала).
В представленном на рис. 11 случае адсорбции ксенона на чистых кристаллах цеолита NaX состава Na2OAl2O33SiO2 газохроматографический метод дает теплоту адсорбции 4,4 ккал/моль, а определение изостерической теплоты адсорбции при нулевом заполнении по уравнению (II, 1), константы которого определены из вакуумных статических измерений, дает 4,5 ккал/моль.
Энтропия адсорбции (изменение дифференциальной энтропии адсорбата при адсорбции на данном адсорбенте) равна
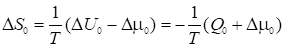
, (34)
Где U0 - соответствующее дифференциальное изменение внутренней энергии адсорбционной системы, a Q0 - дифференциальная теплота адсорбции при нулевом (малом) заполнении поверхности адсорбента. Вводя выражения (32) и (30) в (34), получаем:
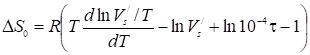
, (35)
Где выражено в см3/м2, a ф в.
Удобно пользоваться величиной
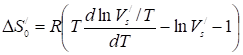
, (36)
В которой выражен в см3/м2. В уравнениях (35) и (36) под знаком логарифма у производной можно использовать также или.
Выше было уже отмечено, что условиям равновесной идеальной хроматографии на однородной поверхности в отсутствие взаимодействия адсорбат-адсорбат отвечают только узкие пики (в идеале оба края пика - передний и задний - должны быть вертикальными, а ширина пика исчезающе малой - малые заполнения поверхности). Поэтому одной симметричности пика, вообще говоря, еще недостаточно для расчетов по равновесным идеальным термодинамическим формулам.
Это можно видеть, в частности, на примере адсорбции нормальных алканов на непористой однородной поверхности графитированной термической сажи и в тонких каналах кристаллов цеолита NaX. В обоих случаях пики симметричны, но с ростом числа атомов углерода в молекуле н. алкана в случае адсорбции в полостях пористых кристаллов цеолита NaX происходит сильное размывание пиков благодаря увеличивающемуся торможению обмена для молекул большого размера. В случае таких сильно размытых, хотя и симметричных, пиков процессы в колонке настолько отклоняются от равновесных, что применять термодинамические формулы для равновесной хроматографии без более глубокого анализа хроматограмм на базе теории неравновесной хроматографии нельзя. Поэтому следует ожидать отклонений термодинамических величин, вычисленных по максимумам сильно размытых симметричных пиков при помощи равновесной теории, от измеренных в статистических условиях. Эти отклонения должны увеличиваться с ростом размеров молекул и энергии адсорбции. На рис. 12 сопоставлены зависимости теплот адсорбции нормальных алканов, определенных газохроматографическим и статическими (из изостер и калориметрических измерений) методами от числа атомов углерода n в молекуле для адсорбции на графитированной термической саже и в полостях пористых кристаллов цеолита NaX.
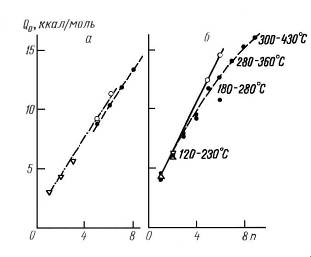
Рис. 22 Зависимость теплот адсорбции Q0 нормальных алканов при малых заполнениях от числачисла атомов углерода n для графитированной термической сажи (а) и кристаллов цеолита (б). Теплоты адсорбции определены статическим (_) - калориметрическим, () - изостерическим), (?) - газохроматографическим методами
В первом случае отклонения невелики и вполне объясняются тем, что газохроматографические опыты для больших n проводились при более высоких температурах, чем статические. В случае же пористых кристаллов оба метода дают совпадающие результаты лишь для низших членов гомологического ряда Результаты газохроматографических определений теплот адсорбции цеолитом для более тяжелых молекул оказываются заниженными.
Случай несимметричных пиков
В этом случае форма пика зависит от формы изотермы адсорбции (рис 1 - 9. Поэтому определение теплот адсорбции по положению максимумов таких пиков при различных температурах даже для одинаковых проб не является правильным, так как максимумы сложных пиков не передают соответствующую изостеру. Возможность расчета теплот адсорбции из пиков различной формы исследовал Карберри. Проанализировав уравнения материального баланса с учетом диффузии для различных уравнений изотерм адсорбции, он нашел, что в случае пиков, соответствующих искривленным изотермам адсорбции, в частности соответствующих изотерме Фрейндлиха, расчет теплот адсорбции непосредственно по максимумам пиков по уравнению (33) не приводит к правильным величинам.
Поэтому из таких сложных пиков следует определить изотермы адсорбции рассмотренных во втором разделе этой главы методом и уже из этих изотерм определить далее обычным путем изостеры (откладывая lg p относительно 1/T для разных а), из наклонов которых можно найти соответствующие величины
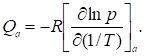
На рис. 13 - 15 представлены полученные из рис. 4, 6 и 7 зависимости Qa от поверхностной концентрации а для адсорбции воды, бензола и метанола на однородной поверхности графитированной термической сажи (или отзаполнения поверхности = а/аm = аm, где am - емкость плотного монослоя, m = 1/ am - площадь, приходящаяся на молекулу адсорбата в этом плотном слое). Из рис. 13 видна близость полученной из хроматограмм (примеры их были даны на рис. 5) зависимости дифференциальной теплоты адсорбции пара бензола от заполнения поверхности этой сажи к соответствующей зависимости, полученной на основании калориметрических измерений.
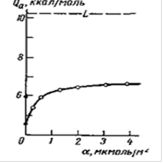
Рис. 23 Зависимость дифференциальной теплоты адсорбции воды от заполнения поверхности графитированной сажи, полученная из хроматограмм рис. 3 и 4. ? - хроматографические данные из работы. L - теплота конденсации
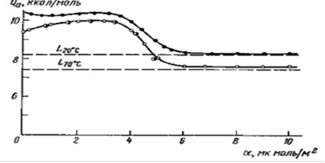
Рис. 24 Зависимость дифференциальной теплоты адсорбции пара бензола от заполнения поверхности графитированной сажи _ - из хроматограмм рис. 5 и изотерм рис. 6; ? - калориметрические данные при 200 С; - значения Qa для 70С, вычисленные из этих калориметрических данных, при помощи значений теплоемкости адсорбированного бензола
В случае адсорбции воды и метанола газохроматографический метод позволил определить весьма низкие величины теплот адсорбции при малых заполнениях, соответствующие взаимодействиям адсорбат - адсорбент (в области малых заполнений не накладывается взаимодействие адсорбат - адсорбат). При больших 6 в этом случае в теплоту адсорбции вносят значительный вклад специфические взаимодействия адсорбат - адсорбат. Это открывает возможность газохроматографического определения энергии водородной связи в монослоях на поверхности неспецифического адсорбента.
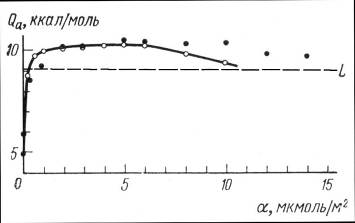
Рис. 25 Зависимость дифференциальной теплоты адсорбции пара метанола от заполнения поверхности графитированной сажи. ? - из хроматограмм и изотерм, _ - из статических измерений
Представляет большой интерес сопоставление газохроматографических данных, полученных при простейшем допущении о практическом достижении адсорбционного равновесия с данными, полученными статическими методами. Некоторые сопоставления такого рода уже были сделаны выше (рис. 12). Он показывает, что на однородных поверхностях оба метода дают близкие результаты. Особенно хорошие результаты получены для однородного непористого и неспецифического адсорбента - графитированной термической сажи. Также близкие к статическим результаты получаются при применении газохроматографического метода к изучению адсорбции относительно слабо адсорбирующихся благородных газов и низших углеводородов на геометрически весьма однородных пористых кристаллах цеолитов (рис. 12). Удовлетворительные результаты были получены также и на неоднородных поверхностях (см., например, рис. 251), однако лишь для сравнительно слабо адсорбирующихся веществ. Худшие результаты получаются обычно для сравнительно сильно адсорбирующихся веществ на адсорбентах с неоднородной поверхностью. Так, было найдено, что полученные газохроматографическим методом теплоты адсорбции нормальных алканов С5--С8 и бензола на силикагелях с порами размером от 100 до 1000 на 15--20% ниже измеренных в калориметре при тех же заполнениях той же поверхности. Такое расхождение может иметь следующие причины: 1) недостижение термодинамического равновесия, 2) влияние предварительной адсорбции воды, аммиака, двуокиси углерода и органических веществ из воздуха во время ввода адсорбента в колонку, 3) влияние адсорбции примесей (например, воды) из газа-носителя, полностью осушить который затруднительно, 4) влияние адсорбции молекул газа-носителя (концентрация которого в колонке велика) на наиболее активных частях поверхности, 5) влияние обычно более высокой температуры в газохроматографических опытах по сравнению со статическими, в частности с калориметрическими опытами и, наконец, 6) влияние ассоциации адсорбат-адсорбат, более сильное в статических опытах при больших заполнениях и более низких температурах.
Вторая и третья причины, т. е. адсорбция воды и других полярных молекул небольшого размера из воздуха до и во время введения адсорбента в колонку и из газа-носителя несущественны S при работе с неспецифическими адсорбентами. Так, из рис. 12 можно действительно видеть, что результаты статических и газохроматографических определений теплот адсорбции нормальных алканов на графитированной саже близки. Эта причина не может быть существенной и при работе со специфическими адсорбентами с очень большой и однородной поверхностью, так как в течение достаточного для ряда хроматографических опытов времени она приводит к отравлению лишь незначительной ее части. Наоборот, при работе с сильными специфическими адсорбентами с низкой удельной поверхностью обе эти причины существенны и действительно приводят к отравлению поверхности достаточно прочно адсорбированными или даже хемосорбированными молекулами. Это наблюдается в случае образцов MgO и ТiO2 с низкой удельной поверхностью. Поэтому изучение адсорбционных свойств чистой поверхности специфических адсорбентов с низкой удельной поверхностью газохроматографическим методом затруднительно, по крайней мере при не очень высоких температурах. Даже для специфических адсорбентов с большой, но неоднородной поверхностью опасность отравления наиболее активных мест адсорбцией примесей существенна и она может стать причиной занижения газохроматографических данных по теплотам адсорбции.
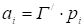
Четвертую причину - влияние адсорбции газа-носителя можно практически устранить, используя в качестве газа-носителя водород или гелий. Шестая причина существенна только при адсорбции молекул группы D на неспецифических адсорбентах при достаточно низких температурах, т. е. скорее в статических опытах; при адсорбции молекул группы D на специфических адсорбентах ее влияние невелико. Таким образом, можно устранить или, по крайней мере, уменьшить неблагоприятное влияние перечисленных факторов, если неспецифические адсорбенты применять при достаточно высоких температурах, а специфические адсорбенты выбирать достаточно однородными и с достаточно высокой удельной поверхностью и применять их также при достаточно высоких температурах.
Для устранения первого из указанных выше неблагоприятных влияний следует применять непористые или макропористые, а также поверхностнопористые адсорбенты. Однако при этом удельная поверхность становится малой, что в случае специфических адсорбентов может привести к сильной адсорбции примесей (воды и т. п.) из воздуха (при заполнении колонки) и из газа-носителя. Сильное же повышение температуры колонки при небольших удельных поверхностях может привести к слишком сильному снижению исправленных времен удерживания, что повлечет за собой снижение точности определений удерживаемых объемов и теплот адсорбции. Таким образом, этот путь вполне надежен лишь для неспецифических адсорбентов.
Влияние неоднородности поверхности и температуры опыта на величины адсорбции и теплот адсорбции следует рассматривать совместно. На рис. 16 приведен пример хроматограмм пропана на силикагеле. Благодаря неоднородности поверхности силикагеля изотермы адсорбции обращены к оси давлений вогнутостью, и поэтому хроматограммы имеют вытянутые хвосты. Однако для различных проб фронты хроматографических полос практически вертикальны, а хвосты сливаются. Таким образом, основной причиной вытянутых хвостов является в данном случае кривизна равновесной изотермы.
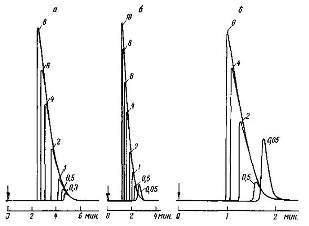
Рис. 26 Хроматограммы пропана на силикагеле при 30°С (а), 50°С (б) и 70°С (в) (колонка 100 х 0,5 см, зернение адсорбента 0,25 - 0,5 мм, скорость газа-носителя - гелия 50 мл/мин, детектор - катарометр) Величины пробы (в мл) указаны у пиков; для пробы 0,05 мл применен пламенно-ионизационный детектор
Вычислив из этих хроматограмм рассмотренным выше методом изотермы адсорбции, можно получить изостеры и определить зависимость теплоты адсорбции от заполнения. Обычно изостеры, полученные из статических измерений, охватывают интервал более низких температур, а изостеры, полученные из хроматограмм, - интервал более высоких температур. При этом возникает существенный вопрос о зависимости теплоты адсорбции от температуры и о влиянии на эту зависимость степени неоднородности поверхности. Ответить на этот вопрос помогает измерение теплоемкости адсорбционных систем. Такие определения сделаны, в частности, для бензола на графитированной термической саже и для н. гексана на силикагеле с удельной поверхностью s = 340 м2/г, и средним диаметром пор d 100 Е.
Теплоемкость адсорбата в обоих случаях оказалась выше теплоемкости соответствующего вещества в газообразном состоянии, следовательно, теплота адсорбции для обоих систем с повышением температуры падает. В обоих случаях теплоемкость адсорбата с ростом заполнения поверхности приближается к теплоемкости жидкости. Однако изменение теплоемкости адсорбата с ростом заполнений на разных адсорбентах происходит различно: теплоемкость адсорбированного бензола на однородной поверхности графитированной термической сажи меньше теплоемкости жидкости и с ростом заполнения растет, а теплоемкость н-гексана на неоднородной поверхности силикагеля больше теплоемкости жидкости и с ростом заполнения падает. Отсюда следует, что теплота адсорбции бензола на однородной поверхности графитированной сажи в области заполнения монослоя уменьшается с повышением температуры медленнее, чем теплота конденсации, а теплота адсорбции н-гексана на неоднородной поверхности силикагеля - быстрее. Теплота адсорбции на неоднородной поверхности падает особенно быстро при малых заполнениях, когда влияние неоднородности поверхности при низких температурах велико.
Таким образом, газохроматографические определения теплот адсорбции, проводимые обычно при температурах более высоких, чем температура опытов, в частности калориметрических, дают при сильной неспецифической адсорбции на однородных поверхностях лишь незначительно заниженные результаты. При адсорбции на очень неоднородных поверхностях газохроматографические определения дают сильно заниженные результаты.
Основной причиной различия газохроматографических и статических определений теплот адсорбции (за исключением специфических адсорбентов с малой поверхностью) является влияние различия температуры в этих определениях, тем более существенное, чем больше неоднородность поверхности адсорбента. Представление об изменении дифференциальной теплоты адсорбции бензола с изменением температуры на однородной поверхности графитированной термической сажи и н-гексана на неоднородной поверхности силикагеля дает рис. 17.
Рис. 27 Хроматограммы пропана на силикагеле при 30°С (а), 50°С (б) и 70°С (в)
Эти результаты показывают, что при достаточно высоких температурах газохроматографический метод дает величины адсорбции и теплот адсорбции, близкие к величинам, полученным статическим методом и приведенным к той же температуре. В этих условиях влияние некоторой неизбежной неравновесности вследствие динамического характера газохроматографического опыта, по-видимому, невелико.
Сильное повышение температуры приводит, однако, к уменьшению разницы в константах равновесия адсорбат - адсорбент (в удерживаемых объемах при малых заполнениях поверхности), т. е. к падению селективности колонки. Поэтому для анализа смесей приходится выбирать некоторую оптимальную температуру или применять программирование температуры колонки, а также адсорбенты с возможно более однородной поверхностью.
Хэбгуд и Ханлан исследовали методом газовой хроматографии адсорбционные свойства ряда активных углей. С этой целью измерялись удерживаемые объемы при разных температурах и определялись теплоты адсорбции. В этой работе было замечено, что величина теплоты адсорбции с изменением температуры несколько меняется. Отмечена также зависимость теплоты адсорбции от заполнения поверхности адсорбента. При небольших пробах веществ величины теплоты адсорбции почти в 2 раза превосходили величину теплоты адсорбции, полученную при больших пробах. Это является результатом того, что на неоднородной поверхности небольшие пробы адсорбируются на самых активных участках поверхности (в тонких порах), где адсорбционный потенциал повышен.
Определение теплот адсорбции различных адсорбатов методом газовой хроматографии позволяет быстро оценить природу поверхности адсорбента.
Из газохроматографических данных были определены теплоты адсорбции низших углеводородов от С1 до С4 на цеолите СаА. Сравнение теплот адсорбции, полученных из газохроматографических данных, с величинами, определенными прямыми калориметрическими измерениями и рассчитанными из изостер по статическим адсорбционным данным, показывает, что метод газовой хроматографии может быть использован для быстрой оценки теплот адсорбции несильно адсорбирующихся газов цеолитами и для исследования их зависимости от строения поверхности адсорбента и молекул адсорбата. Теплоты адсорбции цеолитом нормальных алканов и нормальных алкенов линейно возрастают с увеличением числа атомов углерода в молекуле. При переходе от насыщенных к ненасыщенным соединениям теплоты адсорбции растут в соответствии с энергией дополнительного (к дисперсионному) более специфического взаимодействия преимущественно я электронных связей с катионами поверхности каналов цеолитов.
Это было подтверждено также в работе Киселева, Черненьковой и Яшина, в которой газохроматографическим методом были определены теплоты адсорбции O2, N2, СО и легких углеводородов С1-С4 цеолитами СаА, СаХ и NaX, а также бензола и н. гексана. Теплоты адсорбции азота, этилена и бензола значительно превосходят теплоты адсорбции соответственно кислорода, этана и н-гексана, что объясняется дополнительным вкладом специфического взаимодействия р-электронных связей с катионами поверхности цеолита. В этой работе было замечено также, что по мере увеличения размеров, а также энергии адсорбции молекул углеводородов значения теплот адсорбции цеолитами, определенные газо-хроматографическим методом (при допущении достижения термодинамического равновесия), начинают отставать от значений, определенных статическими методами (рис. 12). Большая энергия адсорбции, а также направленность специфических взаимодействий молекул азота и этилена с находящимися на поверхности цеолитов зарядами приводят к уменьшению относительных (соответственно к метану и пропану) времен удерживания этих молекул цеолитами с повышением температуры. Специфичность адсорбции сильно уменьшается при увлажнении цеолита. Отношение исправленных времен удерживания этилена к соответствующим временам для пропана с увеличением степени влажности цеолита резко падает, тогда как отношение соответствующих времен для этана и пропана практически не зависит от степени влажности цеолита.
В работе также исследовано влияние степени влажности цеолита СаА (5А) навеличины удерживаемых объемов и теплоты адсорбции ряда газов: Н2, O2, N2, СН4, СО, Кг и Хе. Времена удерживания всех газов уменьшаются с увеличением степени гидратации цеолита. Однако это уменьшение происходит более сильно для Н2, О2, N2, CH4 и СО, чем для Кr и Хе; в связи с этим пики Кr и Хе накладываются на соседние пики при изменении количества предварительно адсорбированной цеолитом воды. Теплоты адсорбции, измеренные газохроматографическим способом, с увеличением количества предварительно адсорбированной цеолитом воды повышаются для Кr и Хе, уменьшаются для N2 и СО и остаются практически постоянными для O2 и СН4.
Точность газохроматографического определения теплот адсорбции зависит от точности измерения исправленного времени удерживания, температуры колонки, скорости газа-носителя, давления газа у входа в колонку и т. д. Необходимо отметить, что многие аналитические хроматографы не позволяют с большой точностью определить температуру колонки (во многих хроматографах имеется градиент температуры вдоль колонки), а также не позволяют определить давление у входа в колонку и скорость потока газа. В связи с этим Кнозингером и Спаннхеймером описан хроматограф, предназначенный специально для точных физико-химических исследований при помощи газовой хроматографии. В этом хроматографе можно определять давление у входа в колонку с точностью +0,1 мм рт. ст., скорость потока газа с точностью +0,1 мл/мин, причем градиент температуры в колонке меньше +1°С.
При небольших заполнениях, которые можно осуществить при применении чувствительных детекторов, в особенности ионизационных, взаимодействия адсорбат - адсорбат на однородной поверхности графитированной термической сажи не проявляются, причем не только в случае слабых неспецифических взаимодействий адсорбат - адсорбат, но даже и в случае сильных специфических взаимодействий молекул адсорбата с образованием взаимной водородной связи. Это было показано Беляковой, Киселевым и Ковалевой для адсорбции спиртов на поверхности графитированной термической сажи. Несмотря на некоторое растяжение задней границы пика с ростом числа атомов углерода в молекуле спирта положение максимумов пиков для различных (небольших) доз не изменялось, что указывает на близость изотермы адсорбции при этих малых заполнениях и довольно высоких температурах к изотерме Генри.
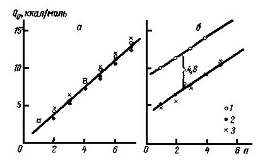
Рис. 28 Зависимость дифференциальных теплот адсорбции нормальных алканов (a) и нормальных спиртов (б) от числа атомов углерода n при малых заполнениях поверхности графитированной термической сажи
На рис. 18 приведены зависимости от числа атомов углерода n величин дифференциальных теплот адсорбции Qa нормальных углеводородов и спиртов, определенных статическими методами при небольших заполнениях (в калориметре и из изостер) и газохроматографическим методом при очень малых заполнениях и более высоких температурах. На этом же рисунке приведены также теоретически вычисленные значения потенциальных энергий адсорбции Ф0 при нулевом заполнении. В случае нормальных алканов дифференциальная теплота адсорбции Qa растет с заполнением монослоя на графитированной саже приблизительно линейно, так что экстраполяцией к нулевому заполнению = 0 легко определить предельное значение Q0 при нулевом заполнении. В случае спиртов зависимость Qa от имеет более сложный вид (рис. 15), так что определить Q0 из калориметрических данных, полученных для более высоких 9, затруднительно. Поэтому на рис. 65 данные статических определений Qa приведены в случае спиртов для 0,1; для нормальных алканов значение Qa при 0,1 весьма близко значению Q0, полученному экстраполяцией к = 0.
Из рис. 18 видно, что для нормальных алканов, молекулы которых взаимодействуют друг с другом только слабо (неспецифически), определения Q0 статическими и газохроматографическими методами дают близкие величины. Эти величины Q0 близки также и к теоретически вычисленным для адсорбции отдельных молекул величинам - Ф0 (на рис. 66 они обозначены крестиками). В случае же спиртов, т. е. молекул группы D, способных специфически взаимодействовать друг с другом, только газохроматографические определения теплот адсорбции (при очень малых заполнениях и повышенных температурах) дали величины Q0, близкие к величинам - Ф0, вычисленным для адсорбции отдельных молекул. Калориметрические же определения, проведенные при значительно больших заполнениях, а также при более низкой температуре (20°С), дали значительно более высокие значения Qa, включающие не только вклад энергии неспецифического взаимодействия адсорбат - адсорбент, но и вклад энергии специфического взаимодействия адсорбат - адсорбат, т. е. энергию взаимной ассоциации молекул спиртов с образованием между ними водородной связи. Действительно, из рис. 18 видно, что разница результатов калориметрических и газохроматографических определений составляет в этом случае 4,8 ккал/моль, т. е. энергию водородной связи между адсорбированными молекулами спиртов.
Этот пример показывает, во-первых, что, комбинируя газохроматографические и калориметрические исследования адсорбции способных к взаимной ассоциации молекул группы D, можно определить энергию водородной связи в адсорбционных слоях на поверхности неспецифических адсорбентов и, во-вторых, при применении графитированных саж и вообще неспецифических адсорбентов первого типа в газохроматографических колонках с чувствительными детекторами и при повышенных температурах благодаря малым заполнениям и снижению взаимодействий адсорбат - адсорбат, можно получить симметричные пики даже в случае веществ, склонных к сильной ассоциации.
При больших величинах проб и более низких температурах колонки с графитированной сажей влияние ассоциации адсорбат - адсорбат можно наблюдать и в газохроматографических опытах. На рис. 6 представлены хроматограммы метанола на графитированной термической саже при различных величинах пробы. Эти хроматограммы имеют сложный вид благодаря влиянию сильного взаимодействия адсорбат - адсорбат и перехода от преимущественно мономолекулярной к преимущественно полимолекулярной адсорбции.
Газохроматографические методы определения удельной поверхности можно разделить на следующие две группы:
- 1) методы вычисления дельной поверхности адсорбентов, основанные на использовании абсолютных величин (относящихся к единице поверхности) удерживаемых объемов Vs, заранее определенных для адсорбентов той же природы с известной удельной поверхностью; 2) методы, основанные на получении изотермы адсорбции изхроматограммы и на расчете из этой изотермы удельной поверхности методом БЭТ.
В первом случае для определения удельной поверхности s можно воспользоваться выражением (23), из которого следует, что
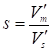
(37)
Где удельный удерживаемый объем для испытуемого адсорбента определяется из хроматографического опыта, а абсолютная величина уже известна.
Эту формулу можно применить только в том случае, если геометрическое или химическое строение поверхности, для которой определена величина, и поверхности, для которой уже известна величина, близки (т. е. только для непористых и достаточно широкопористых адсорбентов одинаковой химической природы), и для близких заполнений (когда величина Fs сильно зависит от заполнения). На рис.31 были показаны примеры зависимости от s для различных адсорбентов. Из которого видно, что пропорциональна s, а наклон прямых равен.
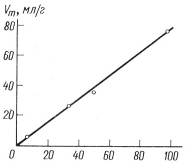
Рис. 29 Зависимость удельного удерживаемого объема н-гексана от величины удельной поверхности s щирокопористых силикагелей (t = 100C)/
Методы определения величины удельной поверхности из удерживаемых объемов впервые были обоснованы в работах. Таким образом, для быстрого определения удельной поверхности s необходимо измерить для достаточно сильно адсорбирующегося пара (чтобы понизить влияние химической неоднородности поверхности адсорбента этот пар должен адсорбироваться неспецифически) на колонке с данной массой адсорбента т и взять из таблиц или определить предварительно значение при той же температуре колонки для адсорбента с той же природой поверхности при близком ее заполнении. Масса адсорбента (длина колонки), адсорбат. величина пробы и температура колонки должны быть выбраны так, чтобы время удерживания можно было бы измерить с достаточной точностью и пики были бы узкими и симметричными.
Второй способ определения удельной поверхности из несимметричных пиков требует определения из хроматограммы равновесной изотермы адсорбции, как описано выше. Из полученных газохроматографическим способом изотерм адсорбции были вычислены удельные поверхности методом БЭТ.
Третий способ определения удельной поверхности, получивший большое распространение, называется методом тепловой десорбции. Этот метод был разработан в 1958 г. Нельсоном и Эггертсоном и Грубнером. Затем он использовался рядом авторов. Метод очень прост и имеет большую чувствительность, что позволяет определять удельные поверхности от 0,01 до 600 м2/г.
Схема измерения по методу тепловой десорбции показана на рис. 19.
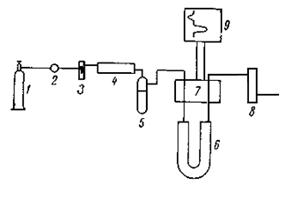
Рис. 30 Схема установки для измерения поверхности адсорбентов по методу тепловой десорбции. 1 - баллон; 2 - вентиль; 3 - ротаметр; 4 - осушитель; 5 - ловушка с цеолитом; 6 - трубка с адсорбентом; 7 - детектор; 8 - измеритель расхода; 9 - самописец
Вентилем тонкой ругулировки 2 устанавливается определенная скорость потока смеси гелия и азота. Поток газа проходит через фильтры 4 и 5, сравнительную ячейку катарометра 7, колонку с исследуемым адсорбентом 6, через измерительную ячейку катарометра и через измеритель расхода газа 8 у выхода из колонки.
После установления равновесия между поверхностью и газом при комнатной температуре колонку с адсорбентом помещают в сосуд Дьюара с жидким азотом. Происходит адсорбция азота на адсорбенте, в результате чего состав газового потока меняется. Это изменение фиксируется детектором. После завершения адсорбции азота состав газа перестает меняться, и перо самописца 9 возвращается к нулевому положению. После этого убирают сосуд Дьюара и быстро нагревают колонку до исходной температуры. Азот при этом десорбируется, состав газа вновь изменяется и на диаграммной ленте самописца записывается пик десорбции (рис. 20). Обычно определение количества адсорбированного при низкой температуре азота ведут по десорбционным выходным кривым, так как они получаются более симметричными (десорбция азота при разогревании до комнатной температуры происходит очень быстро).
Для измерения изотермы адсорбции азота в области перехода к полимолекулярной адсорбции берут обычно смеси с содержанием 5, 10, 15 и 20% азота в гелии.
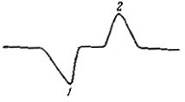
Рис. 31 Пики адсорбции (1) и десорбции (2) азота, полученные при применении метода тепловой десорбции
Вначале проводят калибровку прибора, т. е. определяют зависимость площади пика от концентрации азота. Далее определяют величину адсорбции при различных парциальных давлениях (концентрациях) азота в газе, получают изотерму адсорбции и за
Тем обычным методом БЭТ определяют удельную поверхность адсорбента.
В некоторых случаях нет необходимости снимать всю изотерму адсорбции. Если известна абсолютная величина адсорбции (на единицу поверхности), т. е. поверхностная концентрация монослоя ее, то расчет упрощается. В этом случае легко взять из изотермы значение а, для того значения концентрации азота в газе, для которого проведен опыт по тепловой десорбции. Отношение определенной методом тепловой десорбции величины адсорбции а к абсолютной величине а для того же давления азота в газе прямо дает величину s.
Однако необходимо иметь в виду, что площадь, занимаемая молекулой азота в плотном монослое wm, а следовательно, и поверхностная концентрация в плотном монослое аm = 1/wm (емкость монослоя) довольно сильно зависят от химической природы поверхности адсорбента, так как молекула азота имеет большой квадрупольный момент. В связи с этим Буяновой, Гудковой и Карнауховым было предложено в качестве адсорбата использовать аргон. Площадь, занимаемая молекулой аргона в плотном монослое, принималась равной 15,4 Е2. Для ускорения определения поверхности этими авторами предложена усовершенствованная установка, позволяющая проводить процесс адсорбции одновременно на шести образцах адсорбентов или катализаторов. Установка предусматривает независимую предварительную откачку отдельных образцов и применение интегратора для более быстрого и точного расчета площадей под кривыми десорбции.
При измерениях методом тепловой десорбции необходимо делать поправку на изменение скорости потока при изменении температуры.
С целью дальнейшего уменьшения диффузионного размывания пика было предложено для измерения использовать мелкодисперсный порошок адсорбента. И, наконец, выходную кривую предложено записывать в координатах изменения объемной скорости (регистрируемого детектором, чувствительным к скорости) и времени, так как известно, что при адсорбции и десорбции значительных объемов газа происходит изменение скорости его потока. В этом случае у выхода из колонки вместо детектора концентрации используется регистрирующий измеритель потока. При этом нет необходимости в калибровке детектора.
Куге и Еошикава предложили новый способ измерения удельной поверхности методом газовой хроматографии. Выше была рассмотрена связь между величиной пробы и формой пика. При переходе от адсорбции преимущественно в монослое к преимущественной адсорбции полимолекулярных слоев наблюдалось весьма сложное изменение формы пика при увеличении пробы.
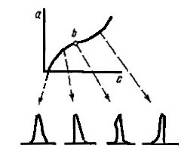
Рис. 32 Изотерма адсорбции
В случае изотермы адсорбции, обращенной выпуклостью к оси заполнения, например изотермы Лэнгмюра, с увеличением пробы времена удерживания уменьшаются. В случае изотермы, обращенной выпуклостью к оси концентрации в газовой фазе, наоборот, с увеличением пробы времена удерживания возрастают (см. рис. 10). Если изотерма адсорбции является комбинацией различных типов, то форма пика имеет вид, показанный на рис. 5, г и д. Авторы работы [32] наблюдали все эти пики при дозировании различных проб в колонку с адсорбентом Сu (Py)2(NO3)2 (рис. 21). При переходе от адсорбции преимущественно монослоем к преимущественно полимолекулярной адсорбции в вершине пика наблюдается излом. Эта точка соответствует точке b на изотерме второго типа по классификации Брунауэра (рис. 6).
Поверхность адсорбента, покрытого плотным монослоем адсорбата, равна:
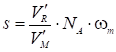
Где V'R - удерживаемый объем адсорбата, соответствующий адсорбции плотного монослоя (еж3); NA - число Авогадро; m - площадь, приходящаяся на одну молекулу адсорбата в плотном монослое; vm -- мольный объем газа при температуре колонки, см3. Величину wm для молекулы адсорбата приближенно можно вычислить по следующей формуле:
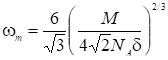
Где - плотность жидкого адсорбата при температуре адсорбции, а М - молекулярный вес. Используя этот метод, авторы работы измерили поверхность адсорбента Сu (Py)2(NO3)2 при применении в качестве адсорбатов бензола, четыреххлористого углерода, циклогексана и н-гексана. Было замечено, что точка излома на пике, соответствующая заполнению монослоя (рис. 21), зависит от скорости потока газа-носителя, температуры колонки и природы адсорбата. Точка излома при скоростях газа, больших, чем 20 см3/мин. По-видимому, при меньших скоростях газа-носителя диффузионное размывание пика маскирует излом в вершине пика. При температурах колонки выше температуры кипения адсорбата подобный излом на пике авторами не наблюдался.
Из газохроматографических данных можно рассчитать не только адсорбционные равновесия, но также вычислить и скорость адсорбции, что очень важно при применении адсорбентов как в самой газовой хроматографии, так и при применении адсорбентов в качестве поглотителей в различных адсорбционных процессах и в качестве катализаторов.
При малых дозах время выхода и форма пиков на хроматограммах отражают адсорбционные взаимодействия адсорбат - адсорбент. Ширина пика определяется различными видами диффузионных процессов, происходящих в колонке с момента ввода пробы до момента ее регистрации детектором у выхода из колонки. Чтобы из хроматограмм вычислить кинетику адсорбции, необходимо отделить размывание, вызванное собственно адсорбционными процессами, от размывания, связанного с процессами диффузии. Точное математическое решение этой задачи очень сложно. Поэтому следует по возможности уменьшить роль диффузионного размывания до такой степени, чтобы им можно было вообще пренебречь, применяя вещества, которые при комнатной температуре адсорбируются очень слабо, как, например, благородные газы и другие низкокипящие и неспецифически адсорбирующиеся вещества. При движении такого газа через адсорбент можно реализовать продольную и "вихревую" диффузию, как и в случае заметно адсорбируемых веществ. Однако при этом процессы адсорбции и десорбции практически исключаются. Чтобы оценить роль диффузионных процессов, время удерживания и форму выходного пика практически неадсорбируемого вещества можно сравнить с временем удерживания и формой пика достаточно сильно адсорбируемого вещества. Экспериментально удобно ввести в колонку одновременно смесь неадсорбируемого и адсорбируемого газов.
В работах Эберли показано, что на колонках небольшой длины процессами внешней диффузии вообще можно пренебречь, В этом случае расчет упрощается и можно получить соотношения, которые позволяют из выходной кривой вычислить кинетику адсорбции.
Особый интерес представляет исследование возможности определения адсорбционных равновесий из неравновесных хромато грамм с учетом диффузионных и кинетических причин размывания хроматографической полосы на проявительной хроматограмме. В работах Грубнера сделана попытка подхода к решению этих важных вопросов.
Похожие статьи
-
Адсорбционные методы исследования свойств поверхности позволяют количественно охарактеризовать происходящие при адсорбции межмолекулярные взаимодействия,...
-
С помощью регистрирующих приборов - самописцев, которые измеряют и автоматически записывают последовательность сигналов детектора, получают кривую...
-
На рис. 8 изображена схема газового хроматографа ХЛ-4. Выходящий из баллона газ-носитель проходит через осушитель 3, дроссель 4, ротаметр 5 и попадает в...
-
Адсорбенты Применяемые адсорбенты характеризуются адсорбционной активностью, истинной и насыпной плотностью, удельной поверхностью общим объемом пор и их...
-
Адсорбция активированный уголь Развитие теории адсорбционных сил еще не достигло такой стадии, когда по известным физико-химическим свойствам газа и...
-
Знаменитая теория полимолекулярной адсорбции Брунауэра, Эммета и Теллера, получившая название теории БЭТ (по первым буквам фамилий ученых), основана на...
-
Попытаемся дать общее представление о свойствах и применении адсорбентов на примере весьма распространенных углеродных материалов. Углеродные адсорбенты...
-
Следует отметить, что не существует особых сил, вызывающих адсорбцию. Адсорбция молекул на поверхности твердого тела происходит за счет сил притяжения со...
-
Описание процессов, происходящих на поверхности, изобилует специальными терминами, и при рассмотрении адсорбционных явлений приходится говорить на языке,...
-
Экспериментальная установка В работе используется прибор для текстурных измерений "Термосорб" серии М, фирмы "КАТАКОН" Серийный №017 Дата выпуска...
-
Теплотой нейтрализации называется количество теплоты, выделенное при взаимодействии 1 моля эквивалента какой-либо кислоты с 1 молем эквивалента...
-
Адсорбция Лэгмюра и Фрейндлиха
Адсорбция. Изотермы адсорбции Лэгмюра, Фрейндлиха. Уравнение БЭТ и его анализ Поверхностная энергия стремится самопроизвольно уменьшиться. Это выражается...
-
Определение поверхности теплопередачи выпарного аппарата Поверхность теплопередачи выпарной установки определяют по основному уравнению теплопередачи:...
-
Адсорбционные явления чрезвычайно широко распространены в живой и неживой природе. Толщи горных пород и почвы являются огромными колоннами с...
-
Общая схема исследования: 1. Составление среднего образца. 2. Извлечение пестицидов из пробы. 3. Очистка экстракта. 4. Анализ экстракта. Прием образцов в...
-
Оборудования, используемые для определения ртути Класс опасности - 1, ПДК в населенных пунктах (среднесуточная) -- 0,0003 мг/мі ПДК в жилых помещениях...
-
Специфические способы определения удельной свободной поверхностной энергии - Коллоидная химия
Поверхностная энергия , энергия, сосредоточенная на границе раздела фаз, избыточная по сравнению с энергией в объеме. При увеличении поверхности раздела...
-
Вычислим значения коэффициента массопередачи в мольных и массовых единицах: (2.7.1) (2.7.2) Подставляя данные в формулы (2.7.1) и (2.7.2) получим: кмоль...
-
Скорость реакции определяется изменением молярной концентрации одного из реагирующих веществ: V=dC/dtV. Факторы, влияющие на скорость химических...
-
Вычисления для следующих входных данных F=1000H m=200 кг m'=1 кг/сек k=2 t0=0 сек V0=0 м/сек B=50 n=50 V1 (t) - результаты, полученные с помощью...
-
Время удерживания (абсолютное) - это отрезок времени, который проходит с момента ввода вещества в колонку до появления максимума пика вещества на...
-
Теплота нейтролизация растворение соль Интегральной теплотой растворения называют теплоту, выделенную или поглощенную при растворении 1 моля вещества в...
-
Q(x) - соответствует площади боковой поверхности данного тела от точки А до точки х. Q(x)>х€[a, x]. Q (x+?x)>х€[a, x+?x], тогда ?Q=Q...
-
Закалочно-испарительный аппарат (Х-1) представляет собой теплообменник смешения. Поток пирогаза охлаждается водой, которая, испаряясь, забирает часть...
-
Плоскость геометрический точка проецирование Длину отрезка АВ можно определить из прямоугольного треугольника АВС-- AС = A1B1...
-
Газовая хроматография - Основы качественного анализа
Этот метод представляет собой замечательное сочетание методов разделения и количественного анализа, поддающееся полной автоматизации. Смесь газов или...
-
Из перечисленного обзора типов ММ, составляющих предмет ИСО, можно выделить следующие особенности ММ ИСО [3]. - Системный подход, заставляющий...
-
Проверка нормальности распределения - Основы научных исследований
Асимметрия и эксцесс позволяют произвести приближенную проверку нормальности распределения. Очевидно, что симметричное и не имеющее эксцесса унимодальное...
-
Класс алкилбензолов представлен 37 соединениями в табл.3, где приведены экспериментальные данные, использованные нами при определении значений параметров...
-
- Ядерная энергетика; - Археология; - Нефтехимия; - Геохимия (изотопная геохронология); - Агрохимия; - Химическая промышленность; - Анализ...
-
Легирование стали повышает ее антикоррозионные свойства. Например, совершенную стойкость к атмосферной коррозии показывают нержавеющие легированные...
-
Раскисление стали - Основы теории окислительной плавки
В чистом железе в равновесии с его оксидами растворяется при температуре 1600?С 0,20...0,23% кислорода. В конце плавке в низкоуглеродистой стали...
-
Электрическое сопротивление - Электрохимические методы исследования
Основной константой, характеризующей электрические свойства вещества, является удельное электрическое сопротивление, зависящее от природы вещества и от...
-
При анализе состава бензиновых фракций широко используют газожидкосую хроматографию. Для получения достоверных результатов хроматографического разделения...
-
Методы выделения - Свойства флавоноидов
Для флавоноидов, как и для других веществ, не существует способа выделения, универсального для всех растительных материалов. В каждом конкретном случае...
-
Вязкость металлов и сплавов - Структура и свойства металлических расплавов
Вязкость, или внутреннее трение, представляет собой внутреннее сопротивление, оказываемое взаимному перемещению смежных слоев жидкости, поэтому и...
-
АНАЛИТИКА Компиляция выдержек из различных источников - удельная б-активность U235 и U238 составляет соответственно 0,08 и 0,012 Бк/мкг (или 80 и 12...
-
Метод сравнения является универсальным методом и применяется во всех разделах статистики (метод сравнения средних, оценивания неизвестных параметров и...
-
Синтез МФ-4 СК партии 29 с применением в качестве восстановителя боргидрида натрия был взят из диссертационной работы Черняевой [18], согласно которой...
-
Задание и исходные данные Необходимо рассчитать насадочную ректификационную колонну для разделения бинарной смеси диоксан - толуол. GD=1000 кг/ч, xF=45%...
Применение газовой хроматографии для исследований адсорбции и определения удельной поверхности твердых тел - Определение теплоты и энтропии адсорбции или растворения на основе хроматографических данных