Характеристика фізико-хімічних показників якості - Формування асортименту і якості цукерок, що реалізуються в місті Білгород-Дністровський
Хімічні методи аналізу базуються на протіканні хімічних реакцій, що супроводжуються помітним зовнішнім ефектом: утворенням осаду чи забарвлених розчинів, виділенням газу, зміною забарвлення об'єкта, що досліджується тощо.
У кількісному аналізі на основі хімічної реакції визначають кількість виділеного продукту реакції, або кількість реактиву, витраченого на утворення цього продукту.
До хімічних методів аналізу відносять Гравіметричний (ваговий) і Титрометричний (об'ємний).
Гравіметричний Метод аналізу базується на визначенні точної маси продукту реакції і малорозчинного осаду. Цей метод є найточнішим, проте в масових аналізах використовується рідко, оскільки є досить громіздким і вимагає багато часу.
При Титрометричному методі вимірюють об'єм витраченого на реакцію розчину реагенту з точно відомою концентрацією. Титрометричний аналіз хоч і менш точний, ніж гравіметричний, але має перевагу в швидкості проведення аналізу.
У товарознавстві найчастіше використовується титрометричний метод.
Застосування хімічних методів у товарознавстві продовольчих товарів
У товарознавстві продовольчих товарів хімічні методи використовуються для визначення вологи, кислотності, азотовмісних речовин (азоту, білку, амінокислот), вітамінів, активності ферментів, жиру тощо.
1. 1. Для визначення вологи застосовують метод азеатропної відгонки та метод Фішера.
Метод Азеатропної відгонки базується на відгонці вологи з високо-киплячою рідиною, що не змішується з водою. Такими речовинами частіше всього є вуглеводні - бензол, толуол, кселол та інші.
Метод застосовують для визначення вологи в пряностях, рибі, сухофруктах та інших товарах.
Метод Фішера базується на взаємодії йоду з діоксидом сірки в розчині метану в присутності води.
- 2. Визначення кислотності (загальної) базується на нейтралізації розчином гідроокису натрію або калію (в присутності індикаторів) водних витяжок вільних кислот з продукту. 3. Визначення Цукрів (вуглеводів) хімічними методами базується на здатності цукрів окислюватися в лужному середовищі. Масову частку цукрів знаходять за кількістю відновлених (встановлених) речовин. Розрізняють наступні хімічні методи визначення цукрів:
- А) встановлення солей окису міді і ртуті в лужному розчині - Перманганатний метод; Б) встановлення в лужному розчині червоної кров'яної солі [K3 Fe (CN)6] - Ферроціанідний метод; В) окислення цукрів, які мають альдегідні групи, йодом у лужному розчині - Йодометричний метод; Г) Колориметричні методи визначення цукрів базуються на здатності цукрів створювати різні стійкі забарвлені речовини. За інтенсивністю забарвлення визначають концентрацію цукрів у розчині товару, що досліджується.
- А) метод К'єльдаля, який базується на виділенні аміаку та уловлюванні його титрованим розчином сірчаної кислоти; Б) біуретовий метод - це здатність пептидних зв'язків білка створити комплексне забарвлення, поєднавшись з іонами міді в лужному розчині; В) метод Лоурі - здатність активних груп різних амінокислот, що входять до складу поліпептидних ланцюгів білка, давати характерні реакції.
Арбітражний метод (метод Сокслета) базується на екстрагуванні жиру з наважки товару розчинником. Далі з одержаної витяжки відганяють розчинник, сирий жир висушують і зважують.
Метод Рушковського С. В. У наважці товару шляхом екстрагування розчинником видаляють жир. Обезжирений залишок висушують і зважують. За різницею між початковою масою речовини і його масою після екстракції визначають кількість жиру в наважці. Наважку продукту настоюють з розчинником у колбі з притертою пробкою. Після цього розчин фільтрують, розчинник відганяють, а залишок висушують і зважують. Це простий метод, але менш точний, ніж інші методи (повна методика визначення жиру цим методом представлена нижче).
- 6. Визначення вітамінів (зокрема вітаміну С, каротину та ін.). 7. Визначення активності ферментів у процесі переробки плодів та овочів.
Нижче представлені методики використання хімічних методів для визначення якості товарів.
Визначення масової частки вологи
Принцип методу полягає в тому, що наважку продукту висушують у сушильній шафі при певній температурі протягом певного часу і за різницею початкової маси і маси сухого залишку установлюють масову частку вологи в продукті.
Як об'єкт дослідження доцільно використовувати різні види і сорти борошна. Вміст вологи в борошні є одним із фізико-хімічних показників якості, що нормується стандартом. До заняття необхідно підготувати не менше 5 зразків пшеничного і житнього борошна.
Для визначення масової частки вологи використовують алюмінієві або скляні бюкси з добре притертими кришками. Бюкси попередньо висушують до постійної маси і зберігають в ексикаторі. Дві наважки досліджуваного зразка масою 5 г (зважені з похибкою не більше 0,01 г), поміщають у висушені, таровані бюкси і висушують протягом 40 хв. при температурі 130°2°С (бюкси з наважкою висушують у відкритому вигляді, кришки ставлять під бюкси). Після 40 хв. бюкси з наважками накривають кришками, виймають із шафи тигельними щипцями і переносять в ексикатор до повного охолодження (приблизно на 15-20 хв.). Після охолодження їх зважують і за різницею між масою наважки до і після висушування визначають масову частку води за формулою, %:
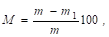
Де M і M1 - маса наважки до і після висушування, г.
Розбіжність між двома паралельними дослідженнями не повинна перевищувати 1 %.
Визначення зольності методом зоління із застосуванням прискорювачів
До заняття готовлять не менше п'яти зразків харчових продуктів із різною зольністю. Рекомендується використовувати харчові продукти, для яких зольність нормується стандартами, наприклад, борошно пшеничне і житнє різних сортів. Дослідні зразки мають бути зашифровані, тобто позначені номерами, які відповідають робочому місцю студентів. Кожний студент індивідуально досліджує зашифрований зразок.
Під час зоління наважки продукту як прискорювач використовують спиртовий розчин оцтовокислого магнію, внаслідок чого зола стає пухкою, або хімічно чисту азотну кислоту, що сприяє окисленню частинок вугілля. Застосування прискорювачів запобігає втратам летких елементів золи (фосфору, калію та ін.), що має місце при високих температурах.
Спиртовий розчин оцтовокислого магнію готують так: 1,61 г оцтовокислого магнію розчиняють у 100 см3 96 %-го етилового спирту, до розчину додають 1-2 кристалики йоду, потім розчин фільтрують.
Проведення аналізу з використанням спиртового розчину оцтовокислого магнію
У тигель з наважкою (борошна, крупи та ін.) вносять піпеткою 3 мл прискорювача і залишають на 1...2 хв. для просочування наважки прискорювачем. Потім тигель переносять під витяжну шафу, ставлять на фарфорову або металеву підставку, наважку запалюють змоченою спиртом ватою, що горить і надіта на металевий стержень. Після згорання спирту тигель поміщають біля дверці відкритої муфельної печі, нагрітої до яскраво-червоного жару. Після вигорання речовин тигель поступово переміщають у глибину муфельної печі. Прожарюють тигель приблизно 1 год. до утворення білої або сірої золи. Потім тигель охолоджують в ексикаторі 35...40 хв. і швидко зважують. Із загальної маси золи вираховують масу золи прискорювача.
Масову частку золи М1 і М2 (на суху і сиру речовину) розраховують за формулами:
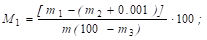
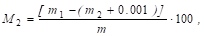
Де m, m1, m2 - відповідно маса наважки продукту, тигля з золою, пустого тигля, г;
M3 - масова частка води в продукті, %;
0,001 - маса оксиду магнію, який утворився із 3 мл доданого спиртового розчину оцтовокислого магнію.
Проведення аналізу з використанням азотної кислоти
Наважку до зоління готують і золять так само, як і при визначенні масової частки золи без прискорювача. Її золять приблизно 1 год. до утворення пухкої маси сірого кольору. Потім тигель виймають із печі і охолоджують на фарфоровій або металевій підставці. Після охолодження в тигель вносять піпеткою або скляною паличкою 2-3 краплі хімічно чистої азотної кислоти, яку випаровують біля відкритої дверці муфельної печі, не допускаючи її кипіння або розбризкування. Після цього тигель поміщають у муфельну піч, нагріту до яскраво-червоного жару. Зоління проводять протягом 20 хв. до повного зникнення із золи темних краплин. Масову частку золи розраховують так, як і в методі без використання прискорювачів. Поправку на прискорювач у формулу не вносять.
Визначення кислотності шкіри
Шкіра та шкіряна тканина завжди містять певну кількість кислоти. Надлишковий вміст мінеральних кислот у шкірі негативно впливає на її властивості, призводить до швидкого руйнування шкіри.
Найбільше поширення одержав потенціометричний метод визначення кислотності шкіри, оснований на вимірюванні рН витяжки, одержаної настоюванням шкіри в 0,1 Н розчині хлориду калію.
Реактиви і матеріали: титрований розчин 0,1 Н NаОН; 0,1 Н розчин хлориду калію; бюретка на 25 мл; піпетки на 10-15 мл; колба з притертим корком місткістю 200 мл; потенціометр; шкіра.
Методика визначення. У колбу з притертим корком поміщають наважку 1,2-1,5 г подрібненої шкіри і додають до неї 50 мл 0,1 Н розчину хлориду калію. Колбу закривають корком і періодично її перемішують протягом чотирьох годин. Після цього розчин відфільтровують і рН - метром вимірюють рН фільтрату шкіряної витяжки.
У випадку необхідності визначення масової частки кислоти одержану хлоркалієву витяжку шкіри можна відтитрувати розчином лугу, графічно знайти точку еквівалентності й обчислити процентний вміст кислоти, наприклад, у перерахунку на сірчану кислоту за формулою:
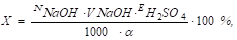
Де N - нормальність титрованого розчину NаОН;
V - об'єм розчину, витраченого на титрування витяжки, мл;
Визначення масової частки жиру за С. В. Рушковським
На лабораторних заняттях масову частку жиру зручно визначати у борошняних кондитерських виробах, наприклад, у печиві. Ці продукти можна швидко підготувати до знежирення, оскільки в них низька масова частка води. Окрім цього, масова частка жиру у печиві є одним з фізико-хімічних показників якості, що нормується стандартами. Бажано для дослідження представити не менше п'яти найменувань печива, виробленого різними кондитерськими фабриками.
Масову частку жиру в досліджуваному продукті визначають за масою знежиреного залишку.
Порядок проведення аналізу. Попередньо подрібнений і висушений продукт масою 1...2 г поміщають у подвійний пакет із фільтрувального паперу, висушеного до постійної маси. Пакети поміщають у бюксу, висушують у сушильній шафі при температурі від 100 до 105 °С до постійної маси. Внутрішній пакет виготовляють із прямокутного аркуша фільтрувального паперу розміром 6 Ч 7 см, зовнішній - розміром 7 Ч 8 см. Внутрішній пакет поміщають у зовнішній таким чином, щоб їх шви не співпадали. Висушені пакети з досліджуваною наважкою поміщають в екстрактор апарату Сокслета, заливають ефіром, збирають апарат (закривають пробкою верхню частину трубки холодильника) і залишають на 8...12 год. Після цього проводять екстрагування до повного витягування жиру з продукту. Об'єм ефіру в прийомній колбі повинен у 1,5 рази перевищувати об'єм екстрактора. Закінчення екстрагування встановлюють так само, як і при прямому методі.
Після витягування жиру пакети виймають з екстрактора, поміщають у бюкси (або на годинникове скло), в яких висушували пакети до екстрагування, і витримують 20-30 хв. у витяжній шафі для усунення ефіру. Потім бюкси з пакетиками висушують у сушильній шафі при температурі від 100 до 105 °С до постійної маси, яку визначають із точністю до 0,001 г.
Масову частку жиру, %, для кожної проби вираховують за формулою:
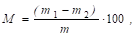
Де M, M1, M2 - відповідно маса наважки продукту, висушеного пакетика до і після екстрагування жиру, г.
Визначення масової частки загального азоту за К'єльдалем
Прилади та обладнання: Установка для спалювання наважки, колби К'єльдаля місткістю 300 см3 із насадкою для спалювання наважки і відгінні колби місткістю 750 см3, конічна колба місткістю 250 см3 (приймач), фарфорова ступка, бюкса і пробірка, аналітичні ваги.
Реактиви: Розчин гідроокису натрію концентрації 0,1 моль/дм3, розчин сірчаної кислоти концентрації 0,1 моль/дм3, концентрована сірчана кислота густиною 1,84; 33 %-й розчин гідроокису натрію, що не містить азотовмісних сполук; хімічно чиста сірчанокисла мідь, метилрот (0,1 г метилроту розчиняють у 30 см3 спирту і розбавляють до 50 см3 Водою), прожарені шматочки пемзи, червоні лакмусові папірці.
Для аналізу беруть таку масу наважки, щоб в ній містилося приблизно 30-40 мг азоту. Маса наважки м'яса не більше 1 г (у середньому 0,5-0,7 г); зерна і борошна - 1-1,5; сирих коренеплодів - 8-10 г; висушених - 1-1,5; сирих бульбоплодів - 8-12 г.
Невелику наважку масою 0,5-0,7 г зважують на аналітичних вагах. У бюксу, в якій є невеличкий шпатель, поміщають 20 г середньої проби дрібно подрібненого продукту, зважують її. Потім відбирають частину проби у пакетик, виготовлений з беззольного фільтру або кальки. Бюксу з продуктами знову зважують і за різницею маси визначають масу наважки, перенесеної у пакетик.
Наважку рослинного продукту масою 1 г зважують у пакетику з кальки. Пакетик з наважкою переносять у чисту суху колбу К'єльдаля таким чином, щоб він потрапив на дно колби. У робочому зошиті записують номер бірки, прикріпленої дротиком до шийки колби.
У витяжній шафі мірним циліндром відмірюють 15-20 см концентрованої сірчаної кислоти густиною 1,84, яку обережно доливають у колбу з наважкою. До неї додають 0,5 г сірчанокислої міді, яка під час наступного нагрівання колби каталізує передачу кисню від сірчаної кислоти до органічних речовин, що окислюються. Колбу у витяжній шафі закріплюють на штативі під нахилом, шийка колби повинна мати насадку. Колбу К'єльдаля нагрівають дуже обережно, оскільки рідина спінюється. Спалювання припиняють, коли вміст колби набуває зеленувато-голубуватого кольору і стає прозорим.
Для відгону аміаку збирають установку, яка відображена на рис. 1.
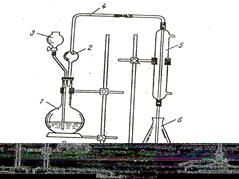
1 - відгінна колба; |
Рис. 1. Установка для відгонки аміаку
Вміст колби охолоджують, потім додають дистильовану воду до 1/3 її об'єму. Воду доливають невеликими порціями по стінках колби. Отриманий розчин без втрат переливають у відгінну колбу. Для запобігання поштовхів під час кипіння в неї додають декілька шматочків (3-4) крупнозернистої пемзи. Реакцію середовища контролюють червоним лакмусовим папірцем. Колбу закривають пробкою, в яку вставлена довга трубка, що відходить від лійки, і коротка трубка, яка відходить від краплеуловлювача. Краплеуловлювач з'єднується з холодильником. З іншого кінця до холодильника приєднують скляну трубку-форштос або алонж, під яким ставлять приймач - конічну колбу, в яку додають з бюретки титрувальної установки 25-30 см3 розчину сірчаної кислоти концентрації 0,1 моль/дм, і додають декілька крапель індикатора. Під час відгонки аміаку реакція середовища в прийомній колбі повинна бути кислою. Нижню частину форштосу занурюють у кислоту.
При включенні холодильника перевіряють міцність і герметичність з'єднання всіх вузлів в установці. Після цього починають відгонку аміаку. У відгінну колбу обережно через лійку доливають із циліндра 100 мл 33 %-го розчину гідроокису натрію. Для запобігання звітрювання аміаку після доливання лугу кран лійки (або зажим Мора, якщо лійка з'єднана гумовою трубкою) закривають. Вміст відгінної колби, який повинен мати лужну реакцію, обережно збовтують. Колбу нагрівають на сітці, спочатку обережно, а потім доводять до кипіння. Аміак разом із парами води потрапляє з відгінної колби через краплеуловлювач у сполучну трубку і холодильник, а з нього безпосередньо в титрований розчин сірчаної кислоти, який завжди беруть із надлишком, щоб запобігти втратам аміаку. У процесі відгону не допускають послаблення нагрівання, оскільки це призводить до втягування кислоти у відгінну колбу. Якщо починається всмоктування кислоти (кислота піднімається до трубки холодильника), трубку, занурену у кислоту, ненадовго виймають із рідини для вирівнювання тиску у приладі. Потім трубку знову занурюють у кислоту і продовжують перегонку.
При нормальному кипінні через 15-20 хв. відганяється 70-90 % аміаку. У прийомну колбу відганяють близько 150 см3 рідини. Повноту відгону аміаку перевіряють червоним лакмусовим папірцем, який змочений дистильованою водою. Форштос віднімають від трубки холодильника і дають краплі відгону стекти на лакмусовий папірець. Якщо колір папірця залишається червоним, відгонку закінчено. Якщо папірець посинів, відгонку необхідно продовжити.
Після відгонки форштос роз'єднують із холодильником, виймають із кислоти, ополіскують дистильованою водою над прийомною колбою і тоді прибирають вогонь з-під відгінної колби. Якщо спочатку загасити пальник, а потім відняти приймач, вміст прийомної колби може потрапити у відгонку через різке падіння тиску.
Залишок кислоти в прийомній колбі відтитровують розчином гідроокису натрію концентрації 0,1 моль/дм3 у присутності метилового оранжевого, чи при подвійному індикаторі метилрот-метиленблау до змінювання забарвлення.
Масову частку загального азоту, %, вираховують за формулою:
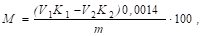
Де V1 - об'єм розчину сірчаної кислоти концентрації 0,1 моль/дм, що знаходиться в прийомній колбі, см3;
К1 - коефіцієнт для перерахунку на розчин кислоти концентрації 0,1 моль/дм3;
V2 - об'єм розчину гідроокису натрію концентрації 0,1 моль/дм, витраченого на титрування надлишку кислоти в прийомній колбі після відгону азоту, см3;
К2 - коефіцієнт для перерахунку на розчин гідроокису натрію (калію) концентрації 0,1 моль/дм3;
M - маса наважки досліджуваного продукту, г;
0,0014 - маса азоту, що відповідає 1 см3 розчину сірчаної кислоти концентрації 0,1 моль/ дм3, г.
Встановивши масу загального азоту, визначають масову частку сирого білку або сирого протеїну в продукті. Для визначення масової частки сирого білка отриману масову частку загального азоту у відсотках множать на відповідний коефіцієнт. Коефіцієнти для перерахунку загального азоту на білок різні тому, що масова частка білка у продуктах неоднакова. Для м'яса, яєць, картоплі й овочів він становить 6,25, для молока і молочних продуктів - 6,37, пшениці, жита, вівса, гороху - 5,68, ячменю, кукурудзи, гречки, квасолі - 6,00.
Визначення вмісту вітаміну С у забарвлених рослинних екстрактах йодометричним методом
Витяжки з рослинного матеріалу титрують розчином йодноватокислого калію в присутності крохмалю як індикатора.
Проведення аналізу. Витяжку для визначення аскорбінової кислоти готують за способом, описаним у завданні 1. Потім відбирають у три конічні колбочки по 10 см3 витяжки, додають по 0,5 см3 1 %-го розчину йодистого калію (КJ), 2 см3 0,5 %-го розчину крохмалю і вміст колбочок відтитровують 0,001 Н розчином йодноватокислого калію (КJО3) до стійкого синього забарвлення. Для того, щоб краще зафіксувати кінець титрування, поруч ставлять колбочку з дослідним екстрактом. При відтитровуванні аскорбінової кислоти від однієї зайвої краплі йодату з'явиться фіолетове забарвлення, яке істотно відрізняється від вихідного забарвлення екстракту.
Паралельно проводять контрольне титрування суміші реактивів, що використовуються в дослідному титруванні (замість досліджуваного фільтрату беруть таку ж саму кількість дистильованої води).
Вміст вітаміну С у продукті (мг на 100 г продукту) розраховують за формулою:
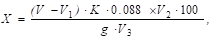
Де V, V1 - об'єм відповідно 0,001н КJО3, що пішло на титрування досліджуваної та контрольної проби, см3;
V2 - загальний об'єм водної витяжки, приготовленої з наважки, см3;
0,088 - кількість мг аскорбінової кислоти, що відповідає 1 см3 0,001н розчину йодноватокислого калію;
К - поправочний коефіцієнт до титру 0,001н розчину КJО3;
G - наважка продукту, г;
V3 - об'єм витяжки, взятої для титрування, см3.
Визначення вмісту каротину у свіжих плодах, овочах і в продуктах їх переробки
Каротин екстрагують із досліджуваного матеріалу ацетоном, потім додають бензин і перемішують. Із бензинового розчину усувають інші каротиноїди (ксантофіл, лікопин та ін.), а також хлорофіл методом хроматографічної адсорбції. Кількість каротину в очищеному бензиновому розчині визначають колориметрично за інтенсивністю жовтого забарвлення розчину порівнянням його з розчином біхромату калію або азобензолу, який стандартизований за чистим каротином.
Проведення аналізу. Оскільки каротин міститься в продуктах рослинного походження, на лабораторному занятті доцільно дослідити вміст каротину у свіжих плодах і овочах та продуктах їх переробки. Особливу увагу слід приділяти методиці підготовки проби свіжих плодів і овочів до аналізу, оскільки різні тканини відрізняються за вмістом каротину. Пробу до аналізу каротину готують так само, як і при дослідженні вітаміну С.
Реактиви: Стандартизований розчин K2Cr2O7 (на аналітичних вагах відважують 0,072 г двічі перекристалізованого K2Cr2O7, розчиняють його в мірній колбі на 100 мл, доводять дистильованою водою до мітки; (1 см3 Цього розчину відповідає 0,0416 мг каротину), ацетон, бензин, вата, кварцевий пісок або бите скло.
Обладнання: Хроматографічна колонка, ділильна лійка, колби Бунзена, лійки Бюхнера, фарфорові ступки, мірні колби місткістю 50 і 100 см3, мірні циліндри, тертушка, фотоелектроколориметр або спектрофотометр, вакуумний насос.
Для аналізу беруть 5-20 г подрібненого на тертушці рослинного матеріалу, ретельно розтирають його в ступці з кварцевим піском або скляним порошком. Оскільки каротин у кислому середовищі нестійкий, то під час розтирання наважки з метою нейтралізації кислот додають трішки соди (Na2СО3). Після попереднього розтирання наважки у ступку наливають 10 мл ацетону та знову розтирають матеріал. Після цього вміст ступки переносять у лійку Бюхнера, яка з'єднана з чистою сухою колбою Бунзена, а остання - з вакуумним або водострумним насосом. На лійку Бюхнера попередньо поміщають два обеззолених фільтри, вирізаних за розміром лійки. Вмикають вакуумний насос і фільтрують. Ступку змивають невеликими порціями ацетону та промивають матеріал на фільтри лійки до повного зникнення забарвлення фільтрату, що стікає. Ацетоновий екстракт переносять у ділильну лійку.
Для того, щоб перевести пігменти в бензин до екстракту, в ділильну лійку додають 10-20 см3 бензину, суміш ретельно перемішують. Ацетон із суміші усувають промивною водою, яку додають невеликими порціями, та легко струшують суміш. Промивні води зливають; вони не повинні містити розчинних у бензині пігментів.
Бензиновому розчину пігментів, який повністю звільнений від ацетону, дають відстоятися від води або висушують його шляхом фільтрування через безводний Na2SО4. Після цього хроматографічною адсорбцією в бензиновому розчині відділяють каротин від хлорофілу, ксантофілу, лікопину та інших пігментів.
На дно хроматографічної колонки щільно вкладають ватний тампон товщиною 1 см, котрий перешкоджає проходженню адсорбента в колбу Бунзена. Потім у колонку вносять невеликими порціями активований МgСО3 або Аl2O3, поступово ущільнюючі кожну порцію скляною паличкою. Довжина стовпчика адсорбенту в колонці повинна становити 5-7 см.
Бензиновий розчин пігментів при слабкому відсмоктуванні пропускають через хроматографічну колонку (необхідно слідкувати за тим, щоб на поверхні постійно був шар бензину, оскільки каротин окислюється під дією повітря). Потім через колонку пропускають чистий бензин доти, доки весь каротин, відділений від інших пігментів, у вигляді жовтої смужки не пройде у прийомну колби. Каротин адсорбується МgСО3 і А12Оз слабше, ніж інші пігменти. Про завершення хроматографування свідчить зникнення жовтого забарвлення елюанта, що витікає з колонки. Бензиновий розчин каротину переносять у мірну колбу на 50 або 100 см3 і доводять бензином до мітки.
Оскільки хімічно чистий каротин є нестійкою речовиною, то під час колориметрування як стандартний розчин використовують водний розчин біхромату калію (К2Сг2О7) або спиртовий розчин азобензолу. 1 см стандартного розчину К2Сг2О7 відповідає 0,00416 мг каротину, 1 см3 стандартного розчину азобензолу - 0,00235 мг каротину.
Вміст каротину в отриманій бензиновій витяжці визначають на фотоелектроколориметрі або спектрофотометрі при довжині хвилі 450 нм. Записують показник екстинції (оптичну густину). На основі виміряної екстинції за побудованою стандартною кривою визначають концентрацію каротину в розчині, а потім вміст каротину (X) у досліджуваному матеріалі в мг на 100 г продукту і розраховують за формулою:
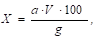
Де А - вміст каротину в 1 см3 розчину, знайденого по графіку, мг;
V - об'єм готового бензинового екстракту каротину, отриманого з наважки, см3;
G - наважка продукту, г.
Вибір методу дослідження якості товарів
Вибір методу дослідження визначається конкретним завданням, яке стоїть перед дослідником. Якщо потрібно визначати основну речовину, наприклад, залізо в сталі або залізній руді чи інші компоненти, що знаходяться у великій кількості, застосовують хімічні методи. Визначення домішок, які містяться у десятих, сотих і тисячних частках відсотка, найкраще проводити високочутливими фізико-хімічними методами аналізу. Роль фізико-хімічних методів особливо зростає у вік прискореного науково-технічного прогресу, коли аналіз того чи іншого об'єкта потрібно провести за короткий час і з досить високою точністю.
Сучасні фізико-хімічні та фізичні методи аналізу широко застосовуються для аналізу високо чистих матеріалів, автоматизації технологічних процесів у різних галузях промисловості, а також для дослідження продовольчих товарів і деяких матеріалів, що йдуть на виробництво непродовольчих товарів.
Похожие статьи
-
Фасують вироби у коробки, пачки або пакети окремими видами або в наборах. Цукерки загорнуті вагові збивні Суфле і лікерні укладають у ящики рядами масою...
-
Основными органолептическими показателями качества конфет являются: вкус и запах, форма, поверхность. Эти показатели различны в зависимости от вида и...
-
В умовах формування ринку продовольчих товарів експертиза має велике значення і свої особливості. Ринкова економіка стимулює розвиток виробництва...
-
Асортимент цукерок дуже різноманітний. Залежно від способу виготовлення і оздоблення поверхні цукерки бувають неглазурованими і глазурованими шоколадною,...
-
Характеристика сировини, яка використовується для виробництва цукерок Цукерки - Дуже велика група кондитерських виробів, які виготовляють на цукровій...
-
1. Ознайомились з факторами формування якості цукерок 2. Провели оцінку якості цукерок за органолептичними показниками 3. Проаналізували фізико-хімічні...
-
Структура асортименту кондитерських виробів супермаркету "Копійка" № п/п Назва Виробник Фасування Ціна за кг. Питома вага, % Вид корпуса 1 Білочка з...
-
Об'єкт та методи дослідження Об'єктами дослідження будуть три види цукерок: - Білочка з горішком - Каракум - Ліщина Назва показника Характеристика за...
-
Вступ - Формування асортименту і якості цукерок, що реалізуються в місті Білгород-Дністровський
В сучасному етапі розвитку економіки важливе місйе займає кондитерська промисловість. В 2004г. объемы потребления сахаристых изделий выросли примерно на...
-
Варіння мильної основи Виготовлення основи туалетного мила із нейтральних жирів періодичним непрямим методом. Цей процес відбувається в котлах 7, 8, 9...
-
Проходячи практику в сільськогосподарському підприємстві ПП АФ "Злагода 21", я визначив що такої посади немає на підприємстві але користуючись додатковою...
-
В результаті тривалої експлуатації валкових млинів з'являються дефекти, які порушують правильний розмел продукту. Найбільш характерні наступні дефекти....
-
Характеристика основної сировини Основною олійною культурою України є соняшник. З нього виробляють більше 75 % рослинної олії від усього виробництва...
-
Виготовлення плодоягідних компотів
Виготовлення плодоягідних компотів Плодоягідні компоти містять вітамін С та Р-вітамінні речовини, особливо якщо сировина відповідає вимогам до сортів та...
-
Актуальность данной темы заключается в том, что мясные баночные консервы пользуются большим спросом у покупателей, являются продуктом, у которого...
-
Для исследования мясных баночных консервов было взято один образец : 1. Свинина тушеная Изготовитель: ООО "Елинский пищевой комбинат", Россия, Московская...
-
Технологическая линия производства йогурта (Дополнение 1) состоит из следующего наименования оборудования: 1. Двухслойный резервуар 3000 л из пищевой...
-
Перед початком проведення ремонтних робіт робочому персоналу необхідно перевірити: А) справність робочого інструменту ( викрутки, пасатижі, кусачки,...
-
Специальные способы литья под давлением - Характеристика литья под давлением
Литье под давлением с использованием вакуума. Для осуществления данного способа литья используют разные методы вакуумирования полости пресс-формы и...
-
Якість продукту -- це сукупність властивостей продукції, яку обумовлюють її придатність, задовольнити певні потреби відповідно до призначення. Від якості...
-
Характеристика и особенности приготовление кальмаров - Блюда из нерыбного морского сырья
Кальмары бывают около 300 видов, распространены широко, преимущественно в тропиках. Кальмары - хищники. Обыкновенный кальмар (Loligo vulgaris) имеет...
-
Холодильная камера холодильника для хранения охлажденной мясной продукциив одноэтажном варианте располагается перед перерабатывающим корпусом....
-
Методы обслуживания потребителей Методы обслуживания потребителей - способ реализации потребителям продукции общественного питания. Различают два метода...
-
Товарозовча характеристика сировини - Технологія приготування пельменів
Борошно - це порошкоподібний продукт, який одержують при розмелюванні хлібних злаків. Назва борошна походить від виду зернової культури. Воно буває:...
-
Характеристика рубленного мяса Мясо содержит много полноценных белков - 14,5 - 23%, жира - от 2 до 37, минеральных веществ - 0,5-1,3% (из них наиболее...
-
Классификация подсолнечного масла Подсолнечное масло имеет характерный приятный аромат и вкус. В зависимости от уровня очистки все масло разделяется на...
-
Управління пральною машиною "Рига-13" полягає в наступному. Зняти кришку машини і встановити віджимний пристрій. Кінець зливного шлангу опустити у...
-
Технічні характеристики по паспорту 1 Найбільша загрузка машини: А) сухої білизни для прання, кг - 1,5; Б) рідини до позначки рівня, л - 28. 2 Час прання...
-
Принцип дії та особливості роботи - Пральна машина "Рига-13". Характеристика, дефекти, ремонт
Прання у побутовій пральній машині типу СМР здіснюється методом циркуляції. Сутність методу заключається у тому, що під дією циркуляції, проникаючи у...
-
Холодные цехи организуются на предприятиях с цеховой структурой производства. Холодные цехи предназначены для приготовления, порционирования и оформления...
-
Мясо хорошо сочетается с различными пищевыми продуктами, поэтому из него можно приготовить большое количество разнообразных блюд. При изготовлении блюд...
-
Заводом "Полимир" запланирована реализация ряда инвестиционных проектов, направленных на реконструкцию существующих производств с целью недопущения...
-
Обзор и характеристика применяемых ГОСТов - Анализ точности и стабильности технологического процесса
Для написания данной курсовой работы были использованы такие ГОСТы как: 1. ГОСТ Р ИСО/ТО 10017-2005 Статистические методы. Руководство по применению в...
-
Основное сырье Колбасные изделия вырабатывают из мяса всех видов скота и птицы, обработанных субпродуктов 1-ой и 2-ой категории, белоксодержащих...
-
Использование технического оборудования снижает трудоемкость первичной обработки сырья, уменьшает процент отходов и т. д. Внедрение механизированных...
-
Характеристики методов получения заготовок Вопросы по теме: Производство заготовок литьем. Производство заготовок пластическим деформированием. Получение...
-
Нефтехимическая промышленность в Республике Беларусь относится к категории стратегических для страны отраслей экономики. Она включает - нефтедобывающий...
-
Зберігають ковбасні вироби, як правило, при температурі не вище 8 О С і 75-80% відносної вологості повітря. Термін реалізації варених ковбас 1 і 2 сорту...
-
Введение, Характеристика готового продукта - Разработка технологии получения строительного материала
В данной курсовой работе "разработка технологии получения строительного материала" мы будем рассматривать следующие решения задач, для достижения цели:...
-
Производство мясных баночных консервов состоит из следующих основных процессов [А. Ф. Шепелев, И. А. Печенежская, 12]: 1. Подготовка сырья (приемка,...
Характеристика фізико-хімічних показників якості - Формування асортименту і якості цукерок, що реалізуються в місті Білгород-Дністровський