Отделение амфотерных металлов от неамфотерных., Отделение меди от свинца. - Подготовка проб к анализу, разделение и концентрирование компонент природных вод
Исследуемый раствор пропускают через колонку с катионитом. При последующем промывании колонки раствором едкой щелочи амфотерные металлы вымываются, неамфотерные остаются на колонке. Таким путем отделяют, например, А13+, Zn2+ от Fe3+, Cu2+ и др..
Отделение меди от свинца.
Катионит в Н-форме задерживает и Си2+ и РЬ2+, при последующем промывании колонки раствором винной кислоты, подщелоченным аммиаком, вымывается свинец, образующий комплексный анион [РЬС4Н2Об]2-. Медь, оставшуюся на колонке в виде катионов [Cu(NH3)4]2+, извлекают затем 5%-ной соляной кислотой.
Метод тонущих частиц. Концентрирование этим способом проводят в большой трубке -- концентраторе (рис), емкостью 1 Л И больше. К оттянутому концу концентратора при помощи резиновой трубки присоединяют пробирку с 10-- 20 Г Влажного катионита. Исследуемой водой, в которой требуется определить содержание примеси никеля, меди, цинка и др. катионов, заполняют весь, концентратор и закрывают пробкой. Под пробкой не должны оставаться пузырьки воздуха. Прибор переворачивают на несколько минут пробкой вниз, пока частицы катионита не пройдут через весь слой жидкости, и затем ставят прибор в первоначальное положение. Катионит медленно опускается и заполняет пробирку. Вся операция отнимает не более 15 Мин. Катионит, извлеченный из пробирки, обрабатывают возможно меньшим объемом теплой 10%-ной соляной кислоты, которая вытесняет сорбированные катионы. Полученный концентрат подвергают дальнейшему анализу.
Для концентрирования катионов применяют катиониты К. У-2 или СБС, измельченные до размера зерен около 0,05--0,2 Мм. Катионит заливают водой и оставляют до следующего дня для набухания. Затем зерна промывают теплой 15%-ной соляной кислотой для удаления примеси железа, никеля и других катионов, промывают несколько раз водой для удаления соляной кислоты. Вместо катионита можно применять несколько граммов измельченного силикагеля, обработанного аммиаком и промытого водой.
Аналогичным методом выделяют рений из природной воды. Воду (500 мл) Подкисляют азотной кислотой и взбалтывают с 0,5 г измельченного активированного угля БАУ. Затем уголь отфильтровывают, промывают разбавленной азотной кислотой, подсушивают и кипятят 2-- 3 мин с 1 н. раствором едкого натра, который извлекает рениевую кислоту. Можно в каплю исследуемого раствора ввести несколько крупинок бесцветного катионита, пропитанных соответствующим реактивом, и наблюдать за окраской крупинок под микроскопом. Более эффективный способ заключается в перемешивании в течение 5 Мин 35--40 набухших зерен катионита СБС или КУ-1 для извлечения катионов или анионита АН-1 или АН-2 для извлечения анионов из нескольких миллилитров исследуемого раствора.
Лучшие результаты получают, если через капиллярную колонку с 35--40 зернами ионита пропустить 10-- 15 мл исследуемого раствора со скоростью 1 мл в 3 мин. Зерна ионита помещают затем на предметное стекло, наносят каплю соответствующего реактива, осаждающего искомый ион в виде малорастворимого кристаллического вещества, и рассматривают под микроскопом. Если считать, что обменная емкость ионита равна только 1 мг-экв/г, То нетрудно вычислить, что одна крупинка ионита (10 -5 Г) Способна поглотить из раствора 10-5 мг-экв, Или в среднем около 10-6 г исследуемого иона, т. е. может практически полностью адсорбировать тот или иной ион, находящийся в очень разбавленном растворе.
Концентрирование на оксицеллюлозе
В качестве ионита применяют также фильтровальную или хроматографическую бумагу, иногда вату, т. е. целлюлозу, представляющую собой полимер состава:
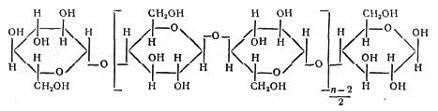
Водородные ионы карбоксильных групп способны обмениваться на катионы металлов. Бумагой нельзя пользоваться для полного выделения и концентрирования катионов из растворов. Однако, обменная емкость материалов из целлюлозы значительно повышается (в 8 -- 10 раз), если ее предварительно окислить, т. е. получить так называемую окси-целлюлозу. Оксиды азота, хлор, бром, другие окислители окисляют целлюлозу. Оксицеллюлозу можно приготовить погружением бумаги в азотную кислоту, после чего бумагу промывают водой. Другой способ получения оксицеллюлозы заключается в промывании гигроскопической ваты 20% - ной соляной кислотой, затем дистиллированной водой. Промытую вату вносят в раствор гипобромита натрия (т. е. раствор NaOH + Br2). Через некоторое время вата расщепляется на тонкие нити, которые промывают водой, затем соляной кислотой и снова водой, после чего высушивают при 90° С. Длина полученных волокон оксицеллюлозы -- 0,5 -- 1 мм, Толщина около 0,01 мм. Оксицеллюлоза очень хороший сорбент для концентрирования многих катионов. Из бумаги, окисленной указанным способом, вырезают диск диаметром 8 мм и закрепляют его в приборе, схема которого показана на рис.
Сначала в кювету наливают небольшой объем раствора карбоната натрия (рН=8). Когда весь раствор пройдет через фильтр, вводят 10 мл природной воды, исследуемой на присутствие небольших количеств никеля. При прохождении воды через фильтр, на последнем сорбируется практически весь никель. Бумажный диск затем извлекают из прибора и погружают в 0,1%-ный этанольный раствор диметилглиоксима, подщелоченный раствором аммиака. При наличии даже 10~9 г никеля во взятых 10 млводы, т. е. при концентрации около 10-9 моль! л, Диск приобретает заметную розовую окраску, по интенсивности которой судят о содержании никеля в пробе.
Анализируемую пробу (1 --10 л) С рН 5--6 взбалтывают с 3 мг мелкоизмельченной оксицеллюлозы и волокна с сорбированными на них ионами, помещают на пористую стеклянную пластинку диаметром 5 мм и отсасывают ( на воронке Бюхнера). Получают диск из волокон, на которых идентифицируют сорбированные ионы при помощи известных цветных реакций. Таким путем удается выделить ряд ионов из проб воды с концентрацией до 1: 109. Указанное количество оксицеллюлозы способно поглотить по 10-5 г Ag+, Cd2+, Ba2+; по 10-6 г А13+, Fe3+, Fe2+, Co2+, Ni2+, Cu2+, Zn2+, Pb2+. Разные ионы задерживаются оксицеллюлозой при разных значениях рН.
|
РН |
Концентрируемые ионы |
|
3 |
Fe3+, Bi3+ |
|
5 |
А13+, Cr3+, Mn2+, Fe2+, Cu2+, VO2+, Cd'2+, Ba2+, UO2+ |
|
6 |
РЬ2+ |
|
8 |
Co2+, Ni2+, Zn2+ |
|
9 |
Ag+, Hg2+ |
Максимальная адсорбция (обмен МеП+) находится при значениях рН, близким к рН осаждения соответствующего гидроксида металла. Для полного извлечения иона иногда требуется повторная обработка раствора свежей порцией оксицеллюлозы. Способность целлюлозы задерживать катионы металлов объясняется не только ионным обменом, но и чисто адсорбционными процессами. Именно поэтому измельченная целлюлоза поглощает большее количество тех или иных катионов, чем неизмельченная.
Катионы, задержанные целлюлозой и оксицеллюлозой, не удаляются промыванием водой, но могут быть выделены обработкой кислотой.
В некоторых случаях нейтральный исследуемый раствор фильтруют через бумагу, пропитанную раствором сульфида цинка, при этом на бумаге задерживаются Cu2+, Pb2+ и другие катионы, которые обнаруживаются по появлению желтой или коричневой окраски.
Концентрирование на стеклянной вате.Этот способ концентрирования основан на том, что на сильноразвитой удельной поверхности стеклянной ваты могут сорбироваться многие ионы. Поверхность 1 Г Стеклянной ваты достигает 2,5 м2 и более.
Исследуемый водный раствор, содержащий ионы железа, подкисляют соляной кислотой (рН<1), обрабатывают перекисью водорода или бромной водой для окисления Fe2+ до Fе3+. Затем добавляют раствор едкой щелочи или аммиака до рН 5--12 и жидкость мед медленно пропускают через колонку (рис.) со стеклянной ватой, предварительно очищенной промыванием соляной кислотой и водой. Железо в виде Fe(OH)3 задерживается ватой. Для растворения Fe(OH)3 через колонку пропускают небольшой объем 0,2 н раствора соляной кислоты. В полученном концентрате определяют железо по реакции с сульфосалициловой кислотой или роданидом аммония.
Аналогичным способом концентрируют и другие элементы.
Выпаривание (упаривание) - Самый простой метод концентрирования, заключающийся в испарении растворителя при нагревании раствора.
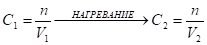
(7)
; V2 V1; С2 С1
N - Количество вещества растворенного компонента, моль,
V1, V2 - объем раствора в начале и конце процесса упаривания.
Применение метода целесообразно только в тех случаях, когда анализируемая вода содержит небольшие количества растворенных анализируемых веществ, не разлагающихся и не улетучивающихся из пробы при нагревании. Такой метод концентрирования применяют, например, при определении содержания тяжелых металлов в питьевой воде, при анализе дождевой воды. В фарфоровой чашке выпаривают 1 - 10 л подкисленной соляной кислотой исследуемой воды приблизительно до 50 - 100 мл, затем катионы осаждают сероводородом или анализируют другими способами. В некоторых случаях объем исследуемой воды достигает 100 л. Несмотря на простоту, этот метод находит ограниченное применение. Исследуемый раствор вместе с малыми количествами определяемых веществ обычно содержит довольно большие количества других соединений. Поэтому выпаривание до очень малых объемов иногда вообще невозможно из-за выделения растворенных веществ. При выпаривании в растворе повышается концентрация всех растворенных веществ, не достигается отделение анализируемого вещества от сопутствующих веществ часто мешающих дальнейшему его определению. Следует учитывать, что при выпаривании возможно загрязнение пробы за счет некоторого растворения материала лабораторной посуды.
Значительно больший интерес представляет выпаривание в сочетании с предварительным экстрагированием. При экстракции отделяется анализируемое вещество от мешающих анализу примесей. Экстракт можно упарить до очень небольшого объема. Если при экстрагировании достигнуто повышение концентрации анализируемого компонента в n раз, а затем при упаривании концентрация повышается еще в m раз, то в общем в результате проведения двух этапов концентрирования увеличение концентрации происходит в nm раз.
Соосаждение - Один из эффективных методов концентрирования (и разделения) анализируемых микрокомпонент из очень разбавленных растворов. Сущность метода заключается в следующем. В разбавленный раствор анализируемого компонента добавляются вещества, при взаимодействии которых друг с другом образуется осадок - Коллектор. Анализируемый компонент ни с одним из добавленных веществ осадка не образует. В процессе осаждения анализируемый микрокомпонент адсорбируется на поверхности частиц образующегося осадка и переходит вместе с ними в осаждаемую твердую фазу - соосаждается. Механизм соосаждения может быть иным (например, могут образовываться смешанные кристаллы и др.). Объем образующегося осадка мал, его растворяют в небольшом количестве соответствующего реагента и анализируют далее каким-либо химическим или физико-химическим методом.
Концентрирование с использованием гидроксида алюминия в качестве коллектора.
В анализируемую пробу воды, загрязненную ионами никеля, добавляют растворимую соль алюминия и гидроксид аммония. Образуется гидроксид алюминия, выпадающий в осадок. Ионы никеля адсорбируются поверхностью гидроксида алюминия, удаляются из раствора с осадком. Процесс протекает при таких значениях рН, при которых гидроксид никеля не образуется
; ; - Коллектор.
Далее осадок отделяется от раствора, растворяется в кислоте, никелиевые соли, которой хорошо растворимы в воде.
При анализе природных вод возможно соосаждение из 1 л воды до
210-6 г Mn2+, Cu2+, Zn2+, Co2+, Ag+, Pb2+ и др.
Концентрирование некоторых ионов путем соосаждения
|
Концентрируемый ион (микрокомпонент) |
Коллектор |
Минимальная концентрация микрокомпонента, При которой возможно его соосаждение |
|
Г/л |
Моль/л |
Применяемый коллектор, кроме способности увлекать с собой микрокомпонент, должен иметь достаточную плотность (для быстрого оседания), хорошую растворимость в кислотах или других растворителях, не мешать определению микрокомпонента к конечном растворе. Желательно, чтобы макрокомпонент удалялся при прокаливании (например, ). Удобными в этом смысле коллекторами являются органические вещества, легкоудаляемые, при прокаливании.
Похожие статьи
-
Возможности подбора и синтеза неорганических сорбентов с заданными свойствами практически неисчерпаемы. Это обусловлено тем, что в качестве сорбентов...
-
Сорбция - Подготовка проб к анализу, разделение и концентрирование компонент природных вод
Сорбцию широко используют для разделения и концентрирования веществ. Сорбционные методы обычно обеспечивают хорошую селективность разделения, высокие...
-
Экстрагируемый Ион Ионы, от которых отделяют Водная фаза Экстрагент 8-оксихинолин хлороформ Метилизобутилкетон и др. Диметилглиоксим, КОН хлороформ и др....
-
Методика отбора и хранения проб Отбор и хранение проб производилось согласно ГОСТ Р 51592-2000 "Общие требования к отбору проб".[35,36] Пробы воды в...
-
Отбор пробы воды следует рассматривать как стадию, в значительной степени определяющую правильность последующего анализа, причем ошибки, допущенные в...
-
Задачи и методы качественного анализа - Основы аналитической химии
Обнаружение или, как иногда говорят, "открытие" отдельных элементов или ионов, входящих в состав веществ - это задачи качественного анализа. Качественный...
-
Предельно допустимая концентрация (ПДК) _ утвержденный в законодательном порядке санитарно-гигиенический норматив. Под ПДК понимается такая концентрация...
-
Проба брал на ГПУ г. Москвы "Природный заказника "Воробъевы горы"из реки Москва, с помощью стеклянной бутыли. Затем стеклянную бутыль опускал в реку и...
-
Общая схема исследования: 1. Составление среднего образца. 2. Извлечение пестицидов из пробы. 3. Очистка экстракта. 4. Анализ экстракта. Прием образцов в...
-
Объемный (титриметрический) анализ, Нейтронно-активационный анализ - Основы качественного анализа
Это один из традиционных химических методов анализа. Для него нужно иметь раствор с точно известной концентрацией вещества, взаимодействующего с...
-
При оценке состояния экосистемы важно учитывать загрязненность водного объекта токсичными веществами. Наибольшую опасность среди них представляют тяжелые...
-
Источники поступления тяжелых металлов (меди) в водоемы Основными загрязнителями окружающей среды являются тяжелые металлы. К ним относятся химические...
-
Что такое гравиметрический фактор F - Основы аналитической химии
Если мы знаем A - навеску анализируемой пробы, b - массу осадка и его состав, то мы можем вычислить содержание определяемого вещества X . X = a*F*100/b...
-
Стандартные методы, как правило, предусматривают использование количественного анализа, позволяющего установить точное содержание отдельных элементов и...
-
Выпаривание пробы воды производится при температуре не более 90-950С на специальной установке, состоящей из электроплитки, песчаной бани, на которую...
-
По существу морская вода является хлоридно-сульфатно-натриево-магниевым раствором, в котором в виде следов и примесей находятся все остальные химические...
-
Задачи и методы количественного анализа - Основы аналитической химии
Количественный анализ - это совокупность химических, физико-химических и физических методов определения количественного соотношения компонентов, входящих...
-
Адсорбционные методы исследования свойств поверхности позволяют количественно охарактеризовать происходящие при адсорбции межмолекулярные взаимодействия,...
-
- Ядерная энергетика; - Археология; - Нефтехимия; - Геохимия (изотопная геохронология); - Агрохимия; - Химическая промышленность; - Анализ...
-
Запасы этих растворов, несоответствующих показателям качества, исчисляются сотнями тысяч тонн и дальнейшее их хранение становится очень опасным для...
-
Особенности анализа органических соединений
Особенности анализа органических соединений: - Реакции с органическими веществами протекают медленно с образованием промежуточных продуктов. -...
-
С помощью регистрирующих приборов - самописцев, которые измеряют и автоматически записывают последовательность сигналов детектора, получают кривую...
-
Основные показатели качества природных вод - Химия воды
Температура -- физический показатель качества воды. Температура воды поверхностных водоисточников в зависимости от сезона года изменяется от 0 до 30°С....
-
АНАЛИТИКА Компиляция выдержек из различных источников - удельная б-активность U235 и U238 составляет соответственно 0,08 и 0,012 Бк/мкг (или 80 и 12...
-
Радиоактивность природных вод В питьевом водоснабжении преимущественно используются поверхностные воды из рек, озер, водохранилищ, а также грунтовые воды...
-
Подготовка угля к коксованию - Анализ и технологическая оценка химического производства
Коксование-- Процесс сухой перегонки каменных углей при их нагревании до 900--1050° С без доступа воздуха. В результате сложных физических и химических...
-
Концентрирование микроэлементов - Природные воды. Классификация примесей
При выборе методов анализа вод различного состава необходимо принимать во внимание приведенные выше данные об элементном составе природных, питьевых и...
-
Физическая сущность методики В связи с отсутствием у нейтронов электрического заряда они проходят в веществе без взаимодействий сравнительно большие...
-
УМЯГЧЕНИЕ И ОБЕССОЛИВАНИЕ ВОДЫ - Химические свойства и строение воды
Под умягчением воды подразумевается процесс удаления из нее катионов жесткости, т. е. кальция и магния. В соответствии с ГОСТ 2874-82 "Вода питьевая"...
-
Диспергированием называют тонкое измельчение твердых мате-риалов или жидкостей и распределение их частиц в жидкой или газообразной среде, в результате...
-
Методы устранения жесткости - Жесткость воды и способы устранения
Чтобы избавиться от временной жесткости необходимо просто вскипятить воду. При кипячении воды, гидрокарбонатные анионы вступают в реакцию с катионами и...
-
Определение содержания железа с помощью фенантролина Методика предназначена для выполнения измерений массовой концентрации общего железа (0,05-2,0...
-
Основным источником поступления ИРН в моря и океаны являются атмосферные выпадения (глобальные и локальные) на их поверхность, а также жидкие стоки...
-
Космогенные радионуклиды поступают в моря и океаны в основном из атмосферы и литосферы, частично могут образовываться в самой водной среде. Их среднее...
-
1. Сушильный шкаф 2. Весы 3. Колонка сит 4. Ступки и пестики 5. Нож, ножницы 6. Металлические или картонные противни 7. Набор счетных геометрий:...
-
Выполнение реакции. В делительную воронку вносят 0,1 мл раствора исследуемого вещества, прибавляют 0,2 мл 0,1%-го раствора родамина 6Ж и 1мл...
-
Индивидуальные признаки катионов 4-й и 5-й аналитических групп - Основы качественного анализа
Многие катионы групп 4 и 5 можно распознать по окраске пламени их летучими соединениями (лучше всего - хлоридами): Na - желтый, Li и Sr - ярко-красные,...
-
Идея электроспрея, хоть и не нова, была возрождена в связи с ее настоящим применением к биомолекулам. Первые эксперименты с электроспреем были проведены...
-
Общие рекомендации к лабораторному практикуму (работам 2-4) - Основы качественного анализа
Чтобы не исказить результаты опытов, пробирки нужно тщательно мыть водопроводной водой и затем ополаскивать небольшим количеством дистиллированной воды....
-
Газовая хроматография - Основы качественного анализа
Этот метод представляет собой замечательное сочетание методов разделения и количественного анализа, поддающееся полной автоматизации. Смесь газов или...
Отделение амфотерных металлов от неамфотерных., Отделение меди от свинца. - Подготовка проб к анализу, разделение и концентрирование компонент природных вод