МЕХАНИЗМ ДЕЙСТВИЯ ФЕРМЕНТОВ - Аминокислоты и ферменты
Структура и функции ферментов, а также механизм их действия почти ежегодно подробно обсуждаются на многих международных симпозиумах и конгрессах. Важное место отводится рассмотрению структуры всей молекулыфермента и ее активных центров, молекулярному механизму действия различных типов ферментов, общей теории энзиматического катализа. Тем не менее до сих пор нет полной ясности по двум кардинальным проблемам энзимологии: чем вызваны специфичность действия и высокая каталитическая эффективность ферментов?
До установления химической природы ферментов гипотезы о механизме их действия опирались на исследования кинетики и модельные опыты химического гомогенного катализа. Повышение скорости химических реакций под действием ферментов объясняли следующим: а) активированием субстрата в результате образования адсорбционных или молекулярных, обратимо диссоциирующих фермент-субстратных комплексов; б) цепныммеханизмом реакций с участием радикалов или возбужденных молекул. Оказалось, что цепные механизмы реакциине играют существенной роли в биологическом катализе. После установления химической природы ферментовподтвердилось представление, выдвинутое более 80 лет назад В. Анри, Л. Михаэлисом и М. Ментен, о том, что при энзиматическом катализе фермент Е соединяется (в принципе обратимо) со своим субстратом S, образуя нестойкий промежуточный фермент-субстратный комплекс ES, который в конце реакции распадается с освобождениемфермента и продуктов реакции Р. Благодаря высокому сродству связывания и образованию ES-комплекса резко возрастает число молекул субстрата, вступающих в реакции. Эти представления легли в основу теории "ключа-замка" Э. Фишера, которую иногда называют теорией "жесткой матрицы". Таким образом, жесткая структураактивного центра оказывается комплементарной молекулярной структуре субстрата, обеспечивая тем самым высокую специфичность фермента.
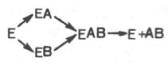
Л. Михаэлис не только постулировал образование промежуточного фермент-субстратного ES-комплекса, но и рассчитал влияние концентрации субстрата на скорость реакции. В процессе реакции различают несколько стадий: присоединение молекулы субстрата к ферменту, преобразование первичного промежуточного соединения в один или несколько последовательных (переходных) комплексов и протекающее в одну или несколько стадий отделение конечных продуктов реакции от фермента. Это можно схематически проиллюстрировать следующими примерами:
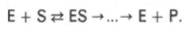
В реакциях анаболизма, например А + В --> АВ, фермент может соединяться как с одним, так и с другим субстратомили обоими субстратами:
В реакциях катаболизма, например АВ --> А + В:
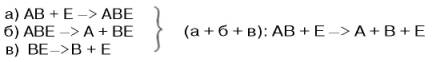
На рис. 4.7 представлена схема образования промежуточного фермент-субстратного комплекса. Если фермент вактивном центре содержит кофермент, то предполагается образование тройного комплекса (рис. 4.8).
Фермент вступает во взаимодействие с субстратом на очень короткий период, поэтому долгое время не удавалось показать образование такого комплекса. Прямые доказательства существования фермент-субстратного комплекса были получены в лабораториях Д. Кейлина и Б. Чанса. В настоящее время экспериментальные и математические методы кинетики, термодинамики и статической механики химических реакций позволяют определить для ряда ферментативных реакций кинетические и термодинамические показатели, в частностиконстанты диссоциации промежуточных фермент-субстратных комплексов, константы скорости и равновесия их образования.
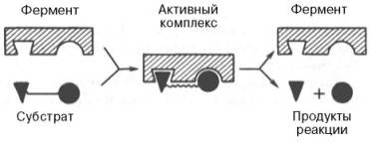
Образование нестойкого фермент-субстратного комплекса согласно теории Э. Фишера "ключ-замок"
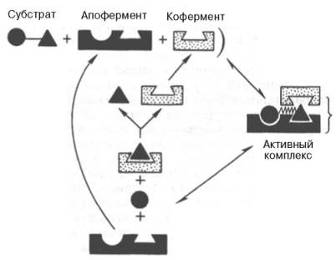
Функция кофер-мента (по А. Кантарову и Б. Шепартцу)
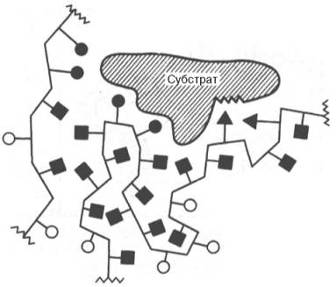
Образование не-ковалентных связей между ферментом и субстратом (схема).
В образовании фермент-субстратных комплексов участвуют водородные связи, электростатические и гидрофобные взаимодействия, а в ряде случаев также ковалентные, координационные связи (рис. 4.9). Информация о природе связей между субстратом и связывающим участком активного центра фермента может быть получена методами ЭПРи ЯМР, а также методами УФ - и ИК-спектроскопии.
Для каталитической активности фермента существенное значение имеет пространственная структура, в которой жесткие участки б-спиралей чередуются с гибкими, эластичными линейными отрезками, обеспечивающими динамические изменения белковой молекулы фермента. Этим изме-неням придается большое значение в некоторых теориях ферментативного катализа. Так, в противоположность модели Э. Фишера "ключ-замок" Д. Кошлендом была разработана теория "индуцированного соответствия", допускающая высокую конформационную лабильностьмолекулы белка-фермента и гибкость и подвижность активного центра. Эта теория была основана на весьма убедительных экспериментах, свидетельствующих о том, что субстрат индуцирует конформационные изменениямолекулы фермента таким образом, что активный центр принимает необходимую для связывания субстратапространственную ориентацию. Иными словами, фермент только в присутствии (точнее, в момент присоединения)субстрата будет находиться в активной (напряженной) Т-форме в отличие от неактивной R-формы (рис. 4.10). На рис. 4.10 видно, что присоединение субстрата S к ферменту Е, вызывая соответствующие изменения конформацииактивного центра, в одних случаях приводит к образованию активного комплекса, в других - неактивного комплекса вследствие нарушения пространственного расположения функциональных групп активного центра в промежуточном комплексе. Получены экспериментальные доказательства нового положения о том, что постулированное Д. Кошлендом "индуцированное соответствие" субстрата и фермента создается не обязательно изменениями конформации белковой молекулы, но также геометрической и электронно-топографической перестройкой молекулысубстрата.
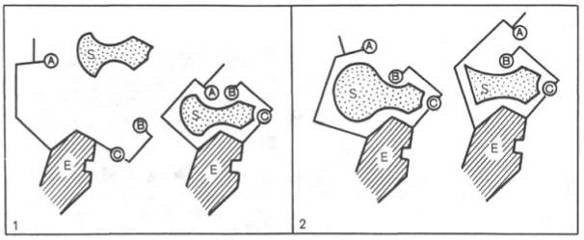
Изменения структуры активного центра фермента, вызванные субстратом, согласно модели "индуцированного соответствия" Д. Кошленда.
А, В, С - функциональные группы активного центра; 1 - активный комплекс; 2 - неактивный комплекс.
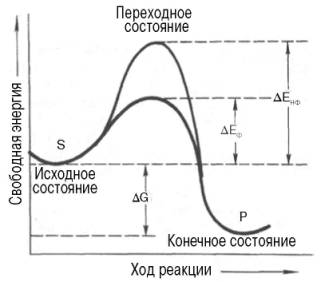
Энергетический механизм ферментативной и неферментативной химических реакций.
S - исходный субстрат; Р - продукт; ДЕНФ - энергия активации неферментативной реакции; ДЕФ - энергия активацииферментативной реакции; ДG - стандартное изменение свободной энергии.
В каталитическом процессе существенное значение имеют точное соответствие между ферментом и субстратом, а также термодинамические и каталитические преимущества подобного соответствия. Гипотеза "индуцированного соответствия" предполагает существование между ферментом и субстратом не только пространственной или геометрической компле-ментарности, но и электростатического соответствия, обусловленного спариванием противоположно заряженных групп субстрата и активного центра фермента. Точное соответствие обеспечивает образование эффективного комплекса между субстратом и ферментом.
Подобно другим катализаторам, ферменты, с термодинамической точки зрения, ускоряют химические реакции за счет снижения энергии активации. Энергией активации называется энергия, необходимая для перевода всехмолекул моля вещества в активированное состояние при данной температуре. Другими словами, это энергия, необходимая для запуска химической реакции, без которой реакция не начинается несмотря на ее термодинамическую вероятность. Фермент снижает энергию активации путем увеличения числа активированныхмолекул, которые становятся реакционноспособными на более низком энергетическом уровне (рис. 4.11). На рисунке видно, что ферментативная реакция имеет более низкую энергию активации. Следует отметить, что как катализируемая ферментом, так и не катализируемая им реакция независимо от ее пути имеет одинаковую величину стандартного изменения свободной энергии (ДG). Действуя на скорость реакции, ферменты не изменяют равновесиямежду прямой и обратной реакциями, как и не влияют на величину свободной энергии реакции; они лишь ускоряют наступление равновесия химической реакции.
Зависимость между константой равновесия и изменением свободной энергии реагирующих веществ математически принято выражать уравнением ДG = = - R*T*lnK, где R - газовая постоянная; Т - абсолютная температура в Кельвинах; lnК - натуральный логарифм константы равновесия; ДG - стандартное изменение свободной энергии, Дж/моль. Из представленного уравнения вытекает, что при высоком значении К величина ДG оказывается отрицательной. Подобные реакции сопровождаются уменьшением свободной энергии. При низком значении К величина ДG оказывается положительной. Если константа равновесия равна единице, то изменение свободной энергии будет равно нулю и реакция легкообратима.
Для измерения константы равновесия и величины свободной энергии какой-либо химической реакции, напримерреакции взаимопревращения глюкозо-1-фосфата в глюкозо-6-фосфат, катализируемой ферментомфосфоглюкомутазой, определяют количество глюкозо-6- и глюкозо-1-фосфата при достижении химического равновесия. В состоянии равновесия содержание глюкозо-6-фосфата оказывается в 19 раз больше количества глюкозо-1-фосфата. Отсюда константа равновесия К равна 19. Подставляя эту цифру в уравнение, получаем ДG = -7329 Дж/моль. Это означает, что при превращении 1 моля глюкозо-1-фосфата в 1 моль глюкозо-6-фосфата притемпературе 25°С происходит уменьшение свободной энергии системы на 7329 Дж.
Таким образом, в механизме ферментативного катализа ведущую роль играют промежуточные фермент-субстратные комплексы, образование которых определяется как тонкой трехмерной структурой активного центра, так и уникальной структурной организацией всей молекулы фермента, обеспечивающими высокую каталитическуюактивность и специфичность действия биокатализатора.
Похожие статьи
-
АКТИВНЫЙ ЦЕНТР ФЕРМЕНТОВ - Аминокислоты и ферменты
При изучении механизма химической реакции, катализируемой ферментами, исследователя всегда интересует не только определение промежуточных и конечных...
-
СТРОЕНИЕ ФЕРМЕНТОВ - Аминокислоты и ферменты
В природе существуют как простые, так и сложные ферменты. Первые целиком представлены полипептидными цепями и при гидролизе распадаются исключительно на...
-
КРАТКАЯ ИСТОРИЯ РАЗВИТИЯ УЧЕНИЯ О ФЕРМЕНТАХ - Аминокислоты и ферменты
Явления брожения и переваривания известны с незапамятных времен, однако зарождение учения о ферментах(энзимология) относится к первой половине XIX в....
-
Высокая каталитическая эффективность. Отличительной особенностью любого фермента является его чрезвычайно высокая каталитическая эффективность. Так,...
-
ПОНЯТИЕ О ФЕРМЕНТАХ - Аминокислоты и ферменты
Аминокислота фермент реакция Ферменты, или энзимы, представляют собой высокоспециализированный класс веществ белковой природы, используемый живыми...
-
Использование предшественников при производстве аминокислот позволяет успешно обходить метаболический контроль, осуществляющийся по механизму обратной...
-
УЛЬТИМОЛЕКУЛЯРНЫЕ ФЕРМЕНТНЫЕ СИСТЕМЫ - Аминокислоты и ферменты
Особую группу ферментов составляют надмолекулярные (или мульти-молекулярные) ферментные комплексы, в состав которых входят не субъединицы (в...
-
Гомогенный и гетерогенный катализ. Механизм действия катализатора - Основы химии
При гомогенном катализе катализатор и реагирующие вещества образуют одну фазу (газовая смесь или раствор). При гетерогенном катализе катализатор и...
-
ИЗОФЕРМЕНТЫ - Аминокислоты и ферменты
Изоферменты, или изоэнзимы,- это множественные формы фермента, катализирующие одну и ту же реакцию, но отличающиеся друг от друга по физическим и...
-
ХИМИЧЕСКАЯ ПРИРОДА ФЕРМЕНТОВ - Аминокислоты и ферменты
В настоящее время получены неопровержимые экспериментальные доказательства белковой природы ферментов. Трудно сейчас представить, что не только Р....
-
Флавинмононуклеотид (ФМН) и флавинадениндинуклеотид (ФАД) функционируют как коферменты для широкого спектра окислительных ферментов и остаются связанными...
-
К-изменение скорости хим. р-ции, под действием особых ве-в-катализаторов, кот. участвуют в ходе р-ции, но к концу ее остаются в неизменном кол-ве. Виды:...
-
Обий механизм Простагландинов много, они по строению мало отличаются друг от друга, но функции их разные. Ряд простагландинов усиливают действие...
-
Характеристика природных и синтетических антиокислителей. Механизм их воздействия Предохранение жиров от порчи имеет важное биологическое и экономическое...
-
Все аминокислоты, из которых состоят белки, являются" L-а-амино - (или имино-) кислотами. Они находят применение как пищевые добавки, приправы, усилители...
-
Механизм действия и биотрансформация - Железо восстановленное (Ferrum reductum)
Фармакокинетика . В присутствии кислоты хлористоводородной желудочного сока железо восстановленное частично превращается в хлорид закиси железа. В...
-
Химическая связь - это взаимное сцепление атомов в молекуле и кристаллической решетке в результате действия между атомами электрических сил притяжения....
-
Как и все органические материалы, полимеры подвержены окислению. Это приводит к изменению вязкости, цвета, охрупчиванию изделий и ухудшению...
-
Літературний огляд, Властивості ферментів - Вплив температури на амілолітичну активність
Властивості ферментів Ферменти - термолабільні сполуки. Це означає, що під дією високих температур вони денатурують. Спочатку при підвищенні температури...
-
Реакции аминокислот - Способы получения аминокислот
Большинство реакций, в которые аминокислоты вступают в лабораторных условиях (in vitro), свойственны всем аминам или карбоновым кислотам. 1. образование...
-
На холоду даже дымящаяся серная кислота (олеум) почти не действует на предельные углеводороды, но при высокой температуре она может их окислять. При...
-
Действие на организм, Получение аммиака - Аммиак
Аммиак сильно раздражает слизистые оболочки уже при 0,5%-ном содержании его в воздухе. Острое отравление аммиаком вызывает поражения глаз и дыхательных...
-
На сегодняшний день существует достаточно большое количество методов моделирования бизнес процессов. Эти методы относятся к разным видам моделирования и...
-
В количественном отношении белки занимают первое место среди всех содержащихся в живой клетке макромолекул; на их долю приходится не менее половины...
-
КАТАЛИЗ В БИОХИМИИ - Процесс катализа
Ферментативный катализ неразрывно связан с жизнедеятельностью организмов растительного и животного мира. Многие жизненно важные химические реакции,...
-
ПРИНЦИП ДЕЙСТВИЯ - Основные понятия о трансформаторах
При подключении к сети в первичной обмотке возникает переменный ток I 1 который создает переменный магнитный поток Ф, замыкающийся по магнитопроводу....
-
Энергия и длина связи. Обменный механизм образования ковалентной связи. Свойства ковалентной связи. Валентность. Донорно-акцепторный механизм образования...
-
Тепловые эффекты при растворении. Механизмы растворения веществ в воде - Растворы
Растворение веществ сопровождается различными тепловыми эффектами в зависимости от природы вещества. При растворении в воде, например, гидроксида калия...
-
При взаимодействии атомао м/у ними может возникнуть хим. связь, приводящая к образованию устойчивой многоатомной с-мы - молекулы, кристалла. Чем прочнее...
-
Действие простагландинов на щитовидную железу. - Простагландины. Строение. Биороль
В щитовидной железе различных животных и в том числе человека, простагландины имитируют многообразные биологические эффекты ТТГ. Простагландины, особенно...
-
Биологическое действие простагландинов - Простагландины. Строение. Биороль
В настоящее время не вызывает сомнений, что простагландины обладают широким спектром фармакологического действия, вызывая при введение изменения почти во...
-
Окисление полиэфиров, Действие света и других излучений - Деструкция полимеров полиэтилентерефталата
Полиэфиры при высоких температурах окисляются под влиянием кислорода воздуха. Ввиду этого реакцию получения полиэфиров проводят в атмосфере азота,...
-
Реакторами идеального (полного) смешения называются реакторы непрерывного действия, в которых осуществляется турбулентный гидродинамический режим. В них...
-
Полупроводниковые квантовые точки могут передавать энергию одному или нескольким подходящим акцепторам [ Clapp A., Medintz I. and Mattoussi H. //...
-
При оценке состояния экосистемы важно учитывать загрязненность водного объекта токсичными веществами. Наибольшую опасность среди них представляют тяжелые...
-
Химические свойства. Образование ковалентной связи по донорно-акцепторному механизму - Аммиак
1. Аммиак - основание Льюиса. Его раствор в воде (аммиачная вода, нашатырный спирт) имеет щелочную реакцию (лакмус - синий; фенолфталеин - малиновый)...
-
Процесс нитрования углеводородов смесью азотной и серной кислот протекает в гетерогенной среде, так как образуются две фазы - органическая...
-
Бизнес-процесс - логически завершенная цепочка взаимосвязанных и повторяющихся видов деятельности, в результате которых ресурсы предприятия используются...
-
Сложение, вычитание, умножение комплексных чисел в алгебраической форме производят по правилам соответствующих действий над многочленами. Четность и...
-
Фармакологическое действие простагландинов и их производных - Простагландины. Строение. Биороль
Эти соединения, по-видимому, вездесущи, их действие сказывается на всех уровнях регуляции физиологических функций. Они способны изменять активность...
МЕХАНИЗМ ДЕЙСТВИЯ ФЕРМЕНТОВ - Аминокислоты и ферменты